Ligandensubstitutionsreaktionen. Komplexe Ioneninstabilitätskonstante, Stabilitätskonstante
Die elementaren Stufen organischer Reaktionen, die durch Säuren, Basen, nukleophile Katalysatoren, Metallkomplexe, feste Metalle und ihre Verbindungen in heterogenen und homogenen Gasphasen- oder Flüssigphasenprozessen katalysiert werden, sind Reaktionen der Bildung und Umwandlung verschiedener organischer und metallorganischer Zwischenprodukte, wie z sowie Metallkomplexe. Organische Zwischenprodukte umfassen Carbeniumionen R + , Carbonium RH 2 + , Carboanionen R–, Anionen- und Kationenradikale, Radikale und Diradikale R , R:, sowie Molekülkomplexe aus organischen Donor- und Akzeptormolekülen (D A ), die sind auch Komplexe mit Ladungstransfer genannt. Bei der homogenen und heterogenen Katalyse durch Metallkomplexe (Metallkomplexkatalyse) organischer Reaktionen sind die Zwischenstufen komplexe (Koordinations-)Verbindungen mit organischen und anorganischen Liganden, metallorganische Verbindungen mit einer MC-Bindung, die in den meisten Fällen Koordinationsverbindungen sind. Ähnlich verhält es sich bei der „zweidimensionalen“ Chemie auf der Oberfläche fester Metallkatalysatoren. Betrachten wir die Hauptreaktionstypen von Metallkomplexen und metallorganischen Verbindungen.
Elementarschritte mit MetallkomplexenDie Reaktionen von Metallkomplexen lassen sich in drei Gruppen einteilen:
a) Elektronentransferreaktionen;
b) Ligandensubstitutionsreaktionen;
c) Reaktionen koordinierter Liganden.
Elektronentransferreaktionen
Bei Elektronentransferreaktionen werden zwei Mechanismen implementiert – der Mechanismus der äußeren Sphäre (ohne Änderungen in den Koordinationssphären von Donor und Akzeptor) und der Mechanismus der Brückenbildung (innere Sphäre), der zu Änderungen in der Koordinationssphäre des Metalls führt.
Betrachten wir den Outer-Sphere-Mechanismus am Beispiel von oktaedrischen Komplexen von Übergangsmetallen. Bei symmetrischen Reaktionen ( G 0 = 0)
Die Geschwindigkeitskonstanten variieren in einem sehr weiten Wertebereich - von 10-12 bis 10 5 l mol-1 sec-1, abhängig von der elektronischen Konfiguration des Ions und dem Grad seiner ᴨȇ-Umstrukturierung während des Prozesses. In diesen Reaktionen manifestiert sich sehr deutlich das Prinzip der geringsten Bewegung - die geringste Änderung in der Valenzschale der Reaktionsteilnehmer.
Bei der Reaktion des Elektronentransfers (1) (Co * ist ein Isotop des Co-Atoms)
(symmetrische Reaktion), Co 2+ (d 7) ᴨȇ geht in Co 3+ (d 6) über. Die elektronische Konfiguration (Valenzschale) ändert sich bei diesem Übergang nicht
6 Elektronen auf dem dreifach entarteten Bindungsniveau bleiben unverändert (), und von der Antibindung e g Ebene wird ein Elektron entfernt.
Geschwindigkeitskonstante zweiter Ordnung für Reaktion (1) k 1 \u003d 1,1 lmol-1 sek-1. Da Phen (Phenanthrolin) ein starker Ligand ist, beträgt die maximale Anzahl 7 d-Elektronen sind gepaart (spingepaarter Zustand). Bei einem schwachen NH3-Liganden ändert sich die Situation dramatisch. Co(NH 3) n 2+ (n = 4, 5, 6) befindet sich im Zustand ohne Spin (High-Spin).
Der stärkere Co(NH 3) 6 3+ -Komplex (~10 30 Mal stärker als Co(NH 3) 6 2+) befindet sich in einem Spin-gepaarten Zustand, ebenso wie der Komplex mit Phen. Dabei muss beim Elektronentransfer die Valenzschale stark umgebaut werden und dadurch k\u003d 10-9 lmol-1 sek-1. Der Umwandlungsgrad von Co 2+ zu Co 3+ von 50 % wird beim Phen-Liganden in 1 Sekunde und beim NH 3 ~ in 30 Jahren erreicht. Offensichtlich kann eine Stufe mit einer solchen Rate (formal elementar) aus der Menge der Elementarstufen bei der Analyse von Reaktionsmechanismen ausgeschlossen werden.
Wert G für die Elektronentransferreaktion während der Bildung eines Stoßkomplexes enthält es gemäß der Marcus-Theorie zwei Komponenten und
Der erste Term ist die Energie der Reorganisation der M-L-Bindungen innerhalb des Komplexes (die Länge und Stärke der Bindung bei einer Änderung des Wertigkeitszustands). Der Wert beinhaltet die Energie der Umlagerung der äußeren Solvathülle im Prozess der Änderung der M-L-Koordinaten und der Ladung des Komplexes. Je kleiner die Änderung der elektronischen Umgebung und je kleiner die Änderung der Länge M-L, desto geringer, je größer die Liganden, desto kleiner und damit desto höher die Elektronentransferrate. Der Wert für den allgemeinen Fall kann mit der Marcus-Gleichung berechnet werden
wo. Bei = 0 .
Im Fall eines Intrasphärenmechanismus wird der Prozess des Elektronentransfers erleichtert, da einer der Liganden des ersten Komplexes einen Brückenkomplex mit dem zweiten Komplex bildet, wodurch einer der Liganden von ihm verdrängt wird
Die Geschwindigkeitskonstanten eines solchen Prozesses sind um 8 Größenordnungen höher als die Konstanten für die Reduktion von Cr(NH 3 ) 6 3+ . Bei solchen Reaktionen muss das Reduktionsmittel ein labiler Komplex sein und der Ligand im Oxidationsmittel muss Brücken bilden können (Cl-, Br-, I-, N 3 -, NCS-, bipy).
LigandensubstitutionsreaktionenEiner der wichtigsten Schritte in der Metallkomplexkatalyse, die Wechselwirkung des Substrats Y mit dem Komplex, verläuft über drei Mechanismen:
a) Ersatz des Liganden durch ein Lösungsmittel. Üblicherweise wird ein solches Stadium als Dissoziation des Komplexes dargestellt
Das Wesentliche des Prozesses ist in den meisten Fällen der Ersatz des Liganden L durch das Lösungsmittel S, das dann leicht durch das Substratmolekül Y ersetzt werden kann
b) Anlagerung eines neuen Liganden entlang einer freien Koordinate unter Bildung eines Assoziats, gefolgt von Dissoziation des substituierten Liganden
c) Synchrone Substitution (Typ S N 2) ohne Bildung einer Zwischenstufe
Bei Pt(II)-Komplexen wird die Reaktionsgeschwindigkeit sehr oft durch die Zweiweggleichung beschrieben
wo k S und k Y sind die Geschwindigkeitskonstanten der Prozesse, die in den Reaktionen (5) (mit Lösungsmittel) und (6) mit Ligand Y ablaufen. Zum Beispiel
Die letzte Stufe des zweiten Wegs ist die Summe dreier schneller Elementarschritte – Spaltung von Cl-, Addition von Y und Eliminierung des H 2 O-Moleküls.
In planaren quadratischen Komplexen von ᴨȇ-Übergangsmetallen wird ein trans-Effekt beobachtet, formuliert von I.I. Für Pt(II)-Komplexe nimmt der trans-Effekt in der Reihe der Liganden zu:
H2O~NH3< Cl- ~ Br- < I- ~ NO 2 - ~ C 6 H 5 - < CH 3 - <
< PR 3 ~ AsR 3 ~ H- < олефин ~ CO ~ CN-.
Das Vorhandensein des kinetischen trans-Effekts und des thermodynamischen trans-Effekts erklärt die Möglichkeit, inerte isomere Komplexe Pt(NH 3) 2 Cl 2 zu synthetisieren:
Reaktionen koordinierter Liganden§ Reaktionen der elektrophilen Substitution (S E) von Wasserstoff durch Metall in der Koordinationssphäre des Metalls und ihre Umkehrprozesse
SH – H 2 O, ROH, RNH 2, RSH, ArH, RCCH.
An solchen Reaktionen sind sogar H 2 - und CH 4 -Moleküle beteiligt
§ Insertionsreaktionen L über M-X-Bindung
Im Fall von X = R (einem metallorganischen Komplex) werden auch metallkoordinierte Moleküle an der M-R-Bindung eingeführt (L - CO, RNC, C 2 H 2 , C 2 H 4 , N 2 , CO 2 , O 2 , etc.). Insertionsreaktionen sind das Ergebnis eines intramolekularen Angriffs des Nucleophils X auf ein vom - oder -Typ koordiniertes Molekül. Rückreaktionen - Reaktionen - und -ausscheidungen
§ Reaktionen der oxidativen Addition und reduktiven Eliminierung
M 2 (C 2 H 2) M 2 4+ (C 2 H 2) 4-
Anscheinend gibt es bei diesen Reaktionen immer eine Vorkoordination des angehängten Moleküls, die aber nicht immer feststellbar ist. In dieser Hinsicht ist das Vorhandensein einer freien Stelle in der Koordinationssphäre oder einer mit einem Lösungsmittel assoziierten Stelle, die leicht durch ein Substrat ersetzt werden kann, ein wichtiger Faktor, der die Reaktivität von Metallkomplexen beeinflusst. Beispielsweise sind Bis-Allyl-Komplexe von Ni gute Vorstufen für katalytisch aktive Spezies, da aufgrund der leichten reduktiven Eliminierung von Bis-Allyl ein Komplex mit einem Lösungsmittel, dem sog. blankes Nickel. Die Rolle freier Plätze wird durch folgendes Beispiel verdeutlicht:
§ Reaktionen der nucleophilen und elektrophilen Addition an - und - Metallkomplexe
Reaktionen metallorganischer VerbindungenAls Intermediate in katalytischen Reaktionen gibt es sowohl klassische metallorganische Verbindungen mit MC-, M=C- und MC-Bindungen als auch nichtklassische Verbindungen, bei denen der organische Ligand gemäß 2, 3, 4, 5 und koordiniert ist 6-Typ oder ist ein Element elektronenarmer Strukturen - überbrückende CH 3 - und C 6 H 6 -Gruppen, nicht klassische Carbide (Rh 6 C (CO) 16, C (AuL) 5 +, C (AuL) 6 2+ , etc.).
Unter den spezifischen Mechanismen für klassische -organometallische Verbindungen stellen wir mehrere Mechanismen fest. Somit wurden 5 Mechanismen der elektrophilen Substitution eines Metallatoms an der M-C-Bindung etabliert.
elektrophile Substitution mit nukleophiler Unterstützung
AdE-Zusatz-Eliminierung
AdE(C)-Bindung an das C-Atom bei sp 2 -Hybridisierung
AdE(M) Oxidative Addition an Metall
Die nukleophile Substitution am Kohlenstoffatom bei den Reaktionen zur Demetallierung metallorganischer Verbindungen erfolgt als Redoxprozess:
Es ist möglich, dass an diesem Schritt ein Oxidationsmittel beteiligt ist.
Als solche Oxidationsmittel können CuCl 2 , p-Benzochinon, NO 3 – und andere Verbindungen dienen. Hier sind zwei weitere elementare Stadien, die für RMX charakteristisch sind:
M-C-Bindungshydrogenolyse
und Homolyse der M-C-Bindung
Eine wichtige Regel, die sich auf alle Reaktionen komplexer und metallorganischer Verbindungen bezieht und mit dem Prinzip der kleinsten Bewegung zusammenhängt, ist Tolmans 16-18-Elektronenschalenregel (Abschnitt 2).
Koordination und metallorganische Verbindungenauf einer OberflächeAn der Oberfläche von Metallen bilden sich nach modernen Vorstellungen Komplexe und metallorganische Verbindungen, ähnlich wie Verbindungen in Lösungen. Für die Oberflächenchemie ist die Beteiligung mehrerer Oberflächenatome an der Bildung solcher Verbindungen und natürlich die Abwesenheit geladener Teilchen wesentlich.
Oberflächengruppen können beliebige Atome (H, O, N, C), Atomgruppen (OH, OR, NH, NH 2 , CH, CH 2 , CH 3 , R), koordinierte Moleküle CO, N 2 , CO 2 , C 2 H 4 , C 6 H 6 . Beispielsweise wurden bei der Adsorption von CO auf der Oberfläche eines Metalls folgende Strukturen gefunden:
Das C 2 H 4 -Molekül auf der Metalloberfläche bildet α-Komplexe mit einem Zentrum und zweifach verknüpften Ethylenbrücken M-CH 2 CH 2 -M, d.h. Im Wesentlichen Metallozyklen
An der Oberfläche von Rh beispielsweise laufen bei der Adsorption von Ethylen mit steigender Temperatur folgende Prozesse der Ethylenumwandlung ab:
Die Reaktionen von Oberflächenzwischenprodukten umfassen die Stufen der oxidativen Addition, reduktiven Eliminierung, Insertion, - und -Eliminierung, Hydrogenolyse von M-C- und C-C-Bindungen und andere Reaktionen des metallorganischen Typs, jedoch ohne das Auftreten freier Ionen. Die Tabellen führen die Mechanismen und Zwischenstufen von Oberflächenumwandlungen von Kohlenwasserstoffen auf Metallen auf.
Tabelle 3.1. Katalytische Reaktionen mit C-C-Bindungsbruch.
Bezeichnungen:
Alkyl, Metallacyclin;
Carben, Allyl;
Karabiner, Vinyl.
Tabelle 3.2. Katalytische Reaktionen unter Bildung einer C-C-Bindung.
Bezeichnungen: siehe Tabelle. 3.1.
Die Bildung aller oben genannten organometallischen Verbindungen auf der Oberfläche von Metallen wurde durch physikalische Methoden bestätigt.
Fragen zur Selbstkontrolle
1) Wie manifestiert sich die Regel der kleinsten Änderung in der Valenzschale eines Metalls während ES in Elektronentransferreaktionen?
2) Warum tragen Koordinationslücken zu einer effektiven Interaktion mit dem Substrat bei?
3) Nennen Sie die wichtigsten Reaktionstypen von koordinierten Liganden.
4) Geben Sie die Mechanismen der elektrophilen Substitution bei den Reaktionen metallorganischer Verbindungen mit HX an.
5) Nennen Sie Beispiele für metallorganische Oberflächenverbindungen.
6) Nennen Sie Beispiele für die Beteiligung von Metallcarben-Oberflächenkomplexen an der Umwandlung von Kohlenwasserstoffen.
Literatur zur Vertiefung
1. Temkin O. N., Kinetik katalytischer Reaktionen in Lösungen von Metallkomplexen, M., MITHT, 1980, Teil III.
2. Kollman J., Higedas L., Norton J., Finke R., Organometallic Chemistry of Transition Metals, M., Mir, 1989, Bd. I, Bd. II.
3. Moiseev I.I., -Complexes in the oxidation of olefins, M., Nauka, 1970.
4. O. N. Temkin, G. K. Shestakov, Yu. A. Treger, Acetylene: Chemistry. Reaktionsmechanismen. Technologie. M., Chemistry, 1991, 416 S., Abschnitt 1.
5. Henrici-Olive G., Olive S., Koordination und Katalyse, M., Mir, 1980, 421 p.
6. O. V. Krylov und V. A. Matyshak, Intermediate Compounds in Heterogeneous Catalysis, Moskau, Nauka, 1996.
7. Zaera F., An Organometallic Guide to the Chemistry of Hydrocarbon Moities on Transition Metal Surfaces., Chem. Soc. Rev., 1995, 95, 2651-2693.
8. Bent B.E., Mimicking Aspects of Heterogeneous Catalysis: Generating, Isolating, and Reacting Proposed Surface Intermediates on Single Crystals in Vacuum, Chem. Soc. Rev., 1996, 96, 1361–1390.
Reaktionen von Koordinationsverbindungen finden immer in der Koordinationssphäre des Metalls mit den darin gebundenen Liganden statt. Es liegt also auf der Hand, dass Liganden in diese Sphäre fallen können müssen, damit überhaupt etwas passieren kann. Dies kann auf zwei Arten geschehen:
- der koordinativ ungesättigte Komplex bindet den neuen Liganden
- in einer bereits abgeschlossenen Koordinationssphäre wechselt ein Ligand zu einem anderen.
Die erste Methode haben wir bereits kennengelernt, als wir uns mit der Koordinations-Ungesättigtheit und der 18-Elektronen-Regel beschäftigten. Lass uns das zweite hier machen.
Liganden jeder Art können in beliebiger Kombination substituiert werden
Aber normalerweise gibt es eine unausgesprochene Regel - die Anzahl der besetzten Koordinationsplätze ändert sich nicht. Mit anderen Worten, die Substitution ändert die Elektronenzahl nicht. Die Substitution eines Liganden eines Typs durch einen anderen ist durchaus möglich und kommt in der Realität häufig vor. Achten wir nur auf die korrekte Handhabung von Ladungen, wenn der L-Ligand zum X-Liganden wechselt und umgekehrt. Wenn wir dies vergessen, ändert sich der Oxidationsgrad des Metalls, und der Austausch von Liganden ist kein Redoxprozess (wenn Sie ein böses Beispiel finden oder finden, lassen Sie es mich wissen - der Offset erfolgt sofort automatisch, wenn Ich kann nicht beweisen, dass Sie sich geirrt haben, warum selbst in diesem Fall garantiere ich einen positiven Beitrag zum Karma).
Substitution mit Haptoliganden
Bei komplexeren Liganden gibt es keine Schwierigkeiten mehr - Sie müssen sich nur an eine ziemlich offensichtliche Regel erinnern: Die Anzahl der Ligandenstellen (dh die Gesamtzahl der Liganden oder Ligandenzentren vom X- oder L-Typ) bleibt erhalten. Dies folgt direkt aus der Erhaltung der Elektronenzahl. Hier sind einige selbstverständliche Beispiele.

Schauen wir uns das letzte Beispiel an. Ausgangsreagenz für diese Reaktion ist Eisendichlorid FeCl 2 . Bis vor kurzem hätten wir gesagt: „Das ist doch nur Salz, was hat die Koordinationschemie damit zu tun?“. Aber solche Ignoranz werden wir uns nicht länger erlauben. In der Chemie der Übergangsmetalle gibt es keine „einfachen Salze“, alle Derivate sind Koordinationsverbindungen, auf die alle Argumente über Elektronenzählung, d-Konfiguration, Koordinationssättigung usw. anwendbar sind. Eisendichlorid, wie wir es zu schreiben gewohnt sind, wäre ein Fe(2+)-Komplex vom Typ MX 2 mit einer d 6 -Konfiguration und 10 Elektronen. Bußgeld? Schließlich haben wir bereits herausgefunden, dass Liganden implizit sind. Um eine Reaktion durchzuführen, benötigen wir ein Lösungsmittel, und für solche Reaktionen ist es höchstwahrscheinlich THF. Die Auflösung des kristallinen Eisensalzes in THF erfolgt gerade dadurch, dass das Donorlösungsmittel freie Plätze einnimmt und die Energie dieses Prozesses die Zerstörung des Kristallgitters kompensiert. Wir wären nicht in der Lage, dieses "Salz" in einem Lösungsmittel aufzulösen, das aufgrund der Lewis-Basizität keine Metallsolvatisierungsdienste bietet. In diesem Fall und in einer Million ähnlicher Fälle ist die Solvatation nur eine Koordinationsinteraktion. Schreiben wir der Deutlichkeit halber das Ergebnis der Solvatation in Form eines FeX 2 L 4 -Komplexes, bei dem zwei Chlorionen in Form von zwei X-Liganden in der Koordinationssphäre verbleiben, obwohl sie höchstwahrscheinlich auch durch verdrängt werden Spender-Lösungsmittelmoleküle mit die Bildung eines geladenen Komplexes FeL 6 2+. In diesem Fall ist es nicht so wichtig. Und so können wir sicher annehmen, dass wir links und rechts einen 18-Elektronen-Komplex haben.
Substitution, Addition und Dissoziation von Liganden sind eng und untrennbar miteinander verbunden
Wenn wir uns an die organische Chemie erinnern, dann gab es zwei Substitutionsmechanismen an einem gesättigten Kohlenstoffatom - SN1 und SN2. Bei der ersten erfolgte die Substitution in zwei Stufen: Der alte Substituent wurde zuerst verlassen und hinterließ ein vakantes Orbital am Kohlenstoffatom, dem ein neuer Substituent mit einem Elektronenpaar folgte. Der zweite Mechanismus ging davon aus, dass der Abflug und die Ankunft gleichzeitig und gemeinsam durchgeführt werden und der Prozess einstufig ist.
In der Chemie der Koordinationsverbindungen kann man sich durchaus etwas Ähnliches vorstellen. Aber es taucht eine dritte Möglichkeit auf, die das gesättigte Kohlenstoffatom nicht hatte – zuerst binden wir einen neuen Liganden an, dann haken wir den alten ab. Es wird sofort klar, dass diese dritte Option kaum möglich ist, wenn der Komplex bereits 18 Elektronen hat und koordinativ gesättigt ist. Aber es ist durchaus möglich, wenn die Anzahl der Elektronen 16 oder weniger beträgt, das heißt, der Komplex ist ungesättigt. Erinnern wir uns sofort an eine offensichtliche Analogie aus der organischen Chemie – die nukleophile Substitution an einem ungesättigten Kohlenstoffatom (in einem aromatischen Ring oder Carbonylkohlenstoff) verläuft ebenfalls zuerst als Addition eines neuen Nukleophils und dann als Eliminierung des alten.
Wenn wir also 18 Elektronen haben, dann läuft die Substitution wie eine Abspaltung ab (Fans von „schlauen“ Wörtern sprechen von dissoziativ-assoziativem oder einfach dissoziativem Mechanismus). Ein anderer Weg würde erfordern, die Koordinationssphäre auf eine Anzahl von 20 Elektronen zu erweitern. Dies ist nicht absolut unmöglich, und solche Optionen werden manchmal sogar in Betracht gezogen, aber es ist definitiv sehr nachteilig und jedes Mal, wenn ein solcher Weg vermutet wird, sind sehr gewichtige Beweise erforderlich. In den meisten dieser Geschichten kamen die Forscher schließlich zu dem Schluss, dass sie etwas übersehen oder nicht berücksichtigt hatten, und der assoziative Mechanismus wurde abgelehnt. Wenn also der ursprüngliche Komplex 18 Elektronen hat, dann muss zuerst ein Ligand gehen, dann soll ein neuer an seine Stelle kommen, zum Beispiel:

Wenn wir einen Hapto-Liganden, der mehrere Positionen besetzt, in die Koordinationssphäre einführen wollen, müssen wir sie zuerst alle freisetzen. Dies geschieht in der Regel nur unter hinreichend strengen Bedingungen, zB um in Chromcarbonyl drei Carbonyle durch η 6 -Benzol zu ersetzen, wird die Mischung viele Stunden unter Druck erhitzt, von Zeit zu Zeit das freigesetzte Kohlenmonoxid abgelassen . Obwohl das Schema die Dissoziation von drei Liganden unter Bildung eines sehr ungesättigten Komplexes mit 12 Elektronen darstellt, erfolgt die Reaktion in Wirklichkeit höchstwahrscheinlich in Stufen, wobei ein Carbonyl zurückbleibt und Benzol in die Kugel eindringt und die Haptizität allmählich durch die Minusstufen erhöht CO - Dihapto - minus eins mehr CO - Tetrahapto - minus eins mehr CO - Hexagapto, so dass weniger als 16 Elektronen nicht erhalten werden.

Wenn wir also einen Komplex mit 16 Elektronen oder weniger haben, dann läuft die Ligandensubstitution höchstwahrscheinlich als Additions-Ablösung ab (für Liebhaber nachdenklicher Worte: assoziativ-dissoziativ oder einfach assoziativ): Erst kommt der neue Ligand, dann der alte Laub. Zwei offensichtliche Fragen stellen sich: Warum geht der alte Ligand, weil 18 Elektronen sehr gut sind, und warum in diesem Fall nicht das Gegenteil tun, wie in 18-Elektronen-Komplexen. Die erste Frage ist leicht zu beantworten: Jedes Metall hat seine eigenen Gewohnheiten, und einige Metalle, insbesondere späte mit fast vollständig gefüllten d-Schalen, bevorzugen die 16-Elektronenzahl und die entsprechenden Strukturtypen und verwerfen daher den zusätzlichen Liganden, der zurückkehrt zu ihrer bevorzugten Konfiguration. Manchmal stört auch der Raumfaktor die Sache, die ohnehin schon vorhandenen Liganden sind groß und der zusätzliche fühlt sich wie ein Buspassagier zur Rush Hour. Es ist einfacher, abzusteigen und zu Fuß herumzulaufen, als so zu leiden. Sie können jedoch einen anderen Passagier hinausschieben, ihn spazieren lassen, und wir gehen. Auch die zweite Frage ist einfach - in diesem Fall müsste der dissoziative Mechanismus erst einen 14-Elektronen-Komplex ergeben, was selten von Vorteil ist.
Hier ist ein Beispiel. Zur Abwechslung ersetzen wir den X-Liganden durch einen L-Liganden und kommen bei den Oxidationsstufen und Ladungen nicht durcheinander. Nochmals: Beim Substituieren ändert sich die Oxidationsstufe nicht, und wenn der X-Ligand weg ist, muss der Verlust durch die Ladung auf dem Metall kompensiert werden. Wenn wir das vergessen, würde die Oxidationsstufe um 1 sinken, was nicht stimmt.

Und noch eine Kuriosität. Aufgrund des freien Elektronenpaars am Stickstoff wurde eine Metall-Pyridin-Bindung gebildet. In der organischen Chemie würden wir in diesem Fall zwangsläufig ein Plus am Stickstoff von Pyridin zeigen (z. B. während der Protonierung oder der Bildung eines quartären Salzes), aber wir tun dies niemals in der Koordinationschemie mit Pyridin oder einem anderen L- Liganden. Das ist für alle, die das strenge und eindeutige System des Strukturzeichnens in der organischen Chemie gewohnt sind, furchtbar ärgerlich, aber man muss sich daran gewöhnen, es ist gar nicht so schwer.
Und es gibt kein genaues Analogon von SN2 in der Chemie der Koordinationsverbindungen, es gibt ein entferntes, aber es ist relativ selten und wir brauchen es nicht wirklich.
Stabile und labile Liganden
Es wäre möglich, überhaupt nicht über die Mechanismen der Ligandensubstitution zu sprechen, wenn da nicht ein äußerst wichtiger Umstand wäre, den wir häufig verwenden werden: Die Substitution von Liganden, ob assoziativ oder dissoziativ, impliziert notwendigerweise die Dissoziation des alten Liganden. Und es ist sehr wichtig für uns zu wissen, welche Liganden leicht und welche schlecht verlassen und lieber in der Koordinationssphäre des Metalls bleiben.
Wie wir bald sehen werden, verbleiben bei jeder Reaktion einige der Liganden in der Koordinationssphäre und ändern sich nicht. Solche Liganden werden normalerweise Zuschauerliganden genannt (wenn Sie solche einfachen, „unwissenschaftlichen“ Wörter nicht wollen, verwenden Sie das englische Wort Zuschauer in der lokalen Transkription Zuschauer, Zuschauerligande, aber ich bitte Sie, kein Zuschauer – das ist unerträglich!) . Und ein Teil nimmt direkt an der Reaktion teil und wird zu Reaktionsprodukten. Solche Liganden werden Schauspieler (nicht Schauspieler!) genannt, also handelnd. Es ist ganz klar, dass die Liganden-Akteure leicht in die Koordinationssphäre des Metalls eingeführt und entfernt werden müssen, sonst bleibt die Reaktion einfach hängen. Aber Zuschauerliganden sollten aus vielen Gründen besser in der Koordinationssphäre belassen werden, aber zumindest aus einem so banalen wie der Notwendigkeit, unnötiges Aufhebens um das Metall zu vermeiden. Es ist besser, wenn nur Liganden, Akteure und in den erforderlichen Mengen, am gewünschten Prozess teilnehmen können. Wenn mehr verfügbare Koordinationsstellen als nötig vorhanden sind, können zusätzliche Liganden-Akteure darauf sitzen und sogar solche, die an Nebenreaktionen teilnehmen, wodurch die Ausbeute des Zielprodukts und die Selektivität verringert werden. Darüber hinaus erfüllen Zuschauerliganden fast immer viele wichtige Funktionen, sorgen beispielsweise für die Löslichkeit von Komplexen, stabilisieren den korrekten Wertigkeitszustand des Metalls, insbesondere wenn es nicht ganz üblich ist, helfen einzelnen Schritten, sorgen für Stereoselektivität usw. Wir entschlüsseln es noch nicht, weil wir das alles ausführlich besprechen werden, wenn wir zu konkreten Reaktionen kommen.
Es stellt sich heraus, dass einige der Liganden in der Koordinationssphäre stark gebunden sein müssen und nicht zur Dissoziation und Substitution durch andere Liganden neigen. Solche Liganden werden genannt koordinativ stabil . Oder einfach stabil, wenn aus dem Kontext klar wird, dass wir über die Bindungsstärke der Liganden sprechen und nicht über deren eigene thermodynamische Stabilität, die uns einfach überhaupt nicht stört.
Und Liganden, die leicht und bereitwillig ein- und austreten und immer bereit sind, anderen Platz zu machen, werden gerufen koordinativ labil , oder einfach labil, und hier gibt es glücklicherweise keine Zweideutigkeiten.
Cyclobutadien als Ligand
Hier ist wohl das schlagendste Beispiel dafür, dass in der Koordinationssphäre ein sehr instabiles Molekül zu einem hervorragenden Liganden werden kann, und per Definition koordinationsstabil, schon deshalb, weil, wenn es sich aus der warmen und kuscheligen Sphäre herauswagt, nichts Gutes erwartet es (auf Kosten der Ausgabe ist nur die Energie der antiaromatischen Destabilisierung).
Cyclobutadien und seine Derivate sind die bekanntesten Beispiele für Antiaromatizität. Diese Moleküle existieren nur bei niedrigen Temperaturen und in stark verzerrter Form - um so weit wie möglich von der Antiaromatizität entfernt zu werden, wird der Zyklus in ein langgestrecktes Rechteck verzerrt, wodurch die Delokalisierung entfernt und die Konjugation von Doppelbindungen so weit wie möglich geschwächt wird (andernfalls , dies nennt man den Jahn-Teller-Effekt der 2. Art: entartetes System, und das Cyclobutadien-Quadrat ist ein entartetes Diradikal, erinnern Sie sich an den Frost-Kreis - verzerrt und reduziert die Symmetrie, um die Entartung zu beseitigen).
Aber in Komplexen sind Cyclobutadien und substituierte Cyclobutadiene ausgezeichnete Tetrahapto-Liganden, und die Geometrie solcher Liganden ist genau ein Quadrat mit identischen Bindungslängen. Wie und warum dies geschieht, ist eine andere Geschichte und bei weitem nicht so offensichtlich, wie es oft dargestellt wird.
Koordinationslabile Liganden
Sie müssen verstehen, dass es zwischen den Bereichen labiler und stabiler Liganden keinen Stahlbetonzaun mit Stacheldraht und Wachtürmen gibt. Erstens hängt es vom Metall ab, und in diesem Zusammenhang funktioniert GMKO gut. Beispielsweise bevorzugen späte Übergangsmetalle weiche Liganden, während frühe Übergangsmetalle harte Liganden bevorzugen. Beispielsweise haftet Iodid sehr fest an den d 8 -Atomen von Palladium oder Platin, tritt aber selten sogar in die Koordinationssphäre von Titan oder Zirkonium in der d 0 -Konfiguration ein. Aber in vielen Metallkomplexen mit nicht so ausgeprägten Merkmalen manifestiert sich Jodid als völlig labiler Ligand, der leicht anderen Platz macht.
Unter sonst gleichen Bedingungen:
- L-Liganden sind im Allgemeinen labiler als X-Liganden;
- die Labilität von X-Liganden wird durch die Härte/Weichheit und Beschaffenheit des Metalls bestimmt;
- „implizite“ Liganden sind sehr labil: Lösungsmittel und Brücken in Dimeren und Clustern, so sehr, dass ihre Anwesenheit in der Koordinationssphäre oft völlig vernachlässigt wird und Strukturen ohne sie mit einer formal ungesättigten Koordinationssphäre gezeichnet werden;
- Digapto-Liganden wie Alkene und Alkine verhalten sich wie typische L-Liganden: Sie sind normalerweise ziemlich labil;
- Liganden mit einer höheren Haptiness sind selten labil, aber wenn ein Polyhapto-Ligand den Bindungsmodus zu einem Monohapto ändern kann, wird er labiler, z. B. verhalten sich η 3 -Allyle so;
- Chelatliganden, die 5- und 6-gliedrige Chelatringe bilden, sind stabil, während Chelate mit weniger oder mehr Ringatomen zumindest an einem Zentrum labil sind (der Chelatring öffnet sich und der Ligand bleibt als einfacher hängen). So verhält sich beispielsweise Acetat;
Koordinationsstabile Liganden
Lass es uns noch einmal machen, aber auf der anderen Seite
In der Koordinationssphäre von Metallen bleiben in der Regel erhalten (sind koordinativ stabil):
- 5- und 6-gliedrige Chelatoren;
- Polyhapto-Liganden: Um Cyclopentadienyle oder Benzole (Arene) aus der Koordinationssphäre zu schlagen, müssen allerlei Spezialtricks angewandt werden – sie kommen einfach nicht so raus, oft halten sie auch längerem Erhitzen stand;
- mit dem Metall assoziierte Liganden mit einem hohen Anteil an der π-Donor-Wirkung (Rückbindung);
- weiche Liganden in späten Übergangsmetallen;
- der „letzte“ Ligand in der Koordinationssphäre.
Die letzte Bedingung sieht seltsam aus, aber stellen Sie sich einen Komplex vor, der viele verschiedene Liganden hat, unter denen es keine unbedingt stabilen gibt (keine Chelatoren und Polygapto-Liganden). Dann ändern sich die Liganden in den Reaktionen relativ gesehen in der Reihenfolge der relativen Labilität. Die am wenigsten labilen und werden die letzten bleiben. Eine solche Fokussierung tritt beispielsweise auf, wenn wir Phosphinkomplexe von Palladium verwenden. Phosphine sind relativ stabile Liganden, aber wenn viele von ihnen vorhanden sind und das Metall reich an Elektronen ist (d 8 , d 10 ), weichen sie nacheinander den Liganden-Aktoren. Der letzte Phosphinligand verbleibt aber meist in der Koordinationssphäre, was im Hinblick auf die Reaktionen, an denen diese Komplexe beteiligt sind, sehr gut ist. Wir werden später auf dieses wichtige Thema zurückkommen. Hier ist ein ziemlich typisches Beispiel, wenn nur ein „letztes“ Phosphan von der anfänglichen Koordinationssphäre des Palladium-Phosphan-Komplexes in der Heck-Reaktion übrig bleibt. Dieses Beispiel bringt uns dem wichtigsten Konzept in Übergangsmetallkomplexreaktionen, dem Konzept der Ligandenkontrolle, sehr nahe. Wir diskutieren später.

Remetallisierung
Wenn ein Ligand durch einen anderen ersetzt wird, ist es wichtig, es nicht mit der Reaktivität des neuen Liganden zu übertreiben. Bei Reaktionen organischer Moleküle ist es uns wichtig, von jedem der Reagenzien genau ein Molekül an die Koordinationssphäre zu liefern. Wenn zwei Moleküle anstelle von einem eintreten, besteht eine hohe Wahrscheinlichkeit von Nebenreaktionen, an denen zwei identische Liganden beteiligt sind. Ein Reaktivitätsverlust ist auch aufgrund der Sättigung der Koordinationssphäre und der Unmöglichkeit, andere für den erwarteten Prozess notwendige Liganden in sie einzuführen, möglich. Dieses Problem tritt besonders häufig auf, wenn starke anionische Nucleophile, z. B. Carbanionen, in die Koordinationssphäre eingeführt werden. Um dies zu vermeiden, werden weniger reaktive Derivate verwendet, bei denen anstelle eines Alkalimetallkations, das eine hohe Bindungs-Ionizität bewirkt, weniger elektropositive Metalle und Halbmetalle (Zink, Zinn, Bor, Silizium etc.) verwendet werden, die kovalente Bindungen ausbilden mit dem nukleophilen Teil . Reaktionen solcher Derivate mit Übergangsmetallderivaten ergeben Ligandensubstitutionsprodukte im Prinzip genau so, als ob das Nucleophil in der anionischen Form wäre, aber aufgrund der verringerten Nucleophilie mit weniger Komplikationen und ohne Nebenreaktionen.
Solche Ligandensubstitutionsreaktionen werden üblicherweise als Transmetallierung bezeichnet, um die offensichtliche Tatsache hervorzuheben, dass das Nucleophil Metalle zu verändern scheint – von elektropositiveren zu weniger elektropositiven. Somit enthält dieser Name ein Element unangenehmer Schizophrenie – wir scheinen uns bereits darauf geeinigt zu haben, dass wir alle Reaktionen aus der Sicht des Übergangsmetalls betrachten, aber plötzlich brechen wir wieder zusammen und betrachten diese Reaktion und nur diese Reaktion aus aus der Sicht eines Nucleophils. Wir müssen uns noch gedulden, so hat sich die Terminologie entwickelt und so wird sie akzeptiert. Tatsächlich geht dieses Wort auf die frühe Chemie metallorganischer Verbindungen zurück und auf die Tatsache, dass die Einwirkung von Lithium- oder Organomagnesiumverbindungen auf die Halogenide verschiedener Metalle und Halbmetalle eine der Hauptmethoden für die Synthese aller metallorganischen, hauptsächlich intransitiven, und die Reaktion, die wir jetzt in der Chemie der Koordinationsverbindungen von Übergangsmetallen betrachten, ist einfach eine Verallgemeinerung der alten Methode der metallorganischen Chemie, aus der alles hervorgegangen ist.
![]()
Wie erfolgt die Remetallisierung?
Remetalling ist sowohl der regulären Substitution ähnlich als auch nicht. Es scheint, dass, wenn wir ein intransitives metallorganisches Reagenz nur als Carbanion mit einem Gegenion betrachten, die Bindung zwischen Kohlenstoff und Übergangsmetall ionisch ist. Aber diese Idee scheint nur für die elektropositivsten Metalle zu gelten - für Magnesium. Aber schon für Zink und Zinn ist diese Vorstellung weit von der Wahrheit entfernt.
Es gehen also zwei σ-Bindungen und vier Atome an ihren Enden in die Reaktion ein. Dadurch werden zwei neue σ-Bindungen gebildet und vier Atome in einer anderen Reihenfolge aneinander gebunden. All dies geschieht höchstwahrscheinlich gleichzeitig in einem viergliedrigen Übergangszustand, und die Reaktion selbst hat einen konzertierten Charakter, wie sehr viele andere Reaktionen von Übergangsmetallen. Die Fülle an Elektronen und Orbitalen für buchstäblich jeden Geschmack und alle Arten von Symmetrien macht Übergangsmetalle in der Lage, gleichzeitig Bindungen in Übergangszuständen mit mehreren Atomen aufrechtzuerhalten.
Bei der Ummetallisierung erhalten wir einen Spezialfall eines sehr allgemeinen Prozesses, den man einfach als σ-Bindungsmetathese bezeichnet. Nicht nur mit echter Metathese von Olefinen und Acetylenen verwechseln, die vollwertige katalytische Reaktionen mit eigenen Mechanismen sind. In diesem Fall sprechen wir über den Mechanismus der Remetallisierung oder einen anderen Prozess, bei dem etwas Ähnliches auftritt.

Liganden - Ionen oder Moleküle, die direkt mit dem Komplexbildner assoziiert sind und Donoren von Elektronenpaaren sind. Diese elektronenreichen Systeme, die über freie und bewegliche Elektronenpaare verfügen, können beispielsweise Elektronendonatoren sein: Verbindungen von p-Elementen weisen komplexbildende Eigenschaften auf und wirken als Liganden in einer Komplexverbindung. Liganden können Atome und Moleküle sein
(Eiweiß, Aminosäuren, Nukleinsäuren, Kohlenhydrate). Die Effizienz und Stärke der Donor-Akzeptor-Wechselwirkung zwischen einem Liganden und einem Komplexbildner wird durch deren Polarisierbarkeit bestimmt, d. h. die Fähigkeit eines Teilchens, seine Elektronenhülle unter äußerem Einfluss umzuwandeln.
Instabilitätskonstante:
Knest= 2 /
K Mund \u003d 1 / Knest
Ligandensubstitutionsreaktionen
Einer der wichtigsten Schritte in der Metallkomplexkatalyse, die Wechselwirkung des Y-Substrats mit dem Komplex, verläuft über drei Mechanismen:
a) Ersatz des Liganden durch ein Lösungsmittel. Üblicherweise wird ein solches Stadium als Dissoziation des Komplexes dargestellt
Das Wesentliche des Prozesses ist in den meisten Fällen der Ersatz des Liganden L durch das Lösungsmittel S, das dann leicht durch das Substratmolekül Y ersetzt werden kann
b) Anlagerung eines neuen Liganden entlang einer freien Koordinate unter Bildung eines Assoziats, gefolgt von Dissoziation des substituierten Liganden
c) Synchrone Substitution (Typ S N 2) ohne Bildung einer Zwischenstufe
Vorstellungen über die Struktur von Metalloenzymen und anderen Biokomplexverbindungen (Hämoglobin, Cytochrome, Cobalamine). Physikalische und chemische Prinzipien des Sauerstofftransports durch Hämoglobin.
Strukturmerkmale von Metalloenzymen.
Biokomplexverbindungen unterscheiden sich beträchtlich in ihrer Stabilität. Die Rolle des Metalls in solchen Komplexen ist sehr spezifisch: Schon der Ersatz durch ein Element mit ähnlichen Eigenschaften führt zu einem signifikanten oder vollständigen Verlust der physiologischen Aktivität.
1. B12: enthält 4 Pyrrolringe, Kobaltionen und CN-Gruppen. Fördert die Übertragung des H-Atoms auf das C-Atom im Austausch gegen eine beliebige Gruppe, beteiligt sich an der Bildung von Desoxyribose aus Ribose.
2. Hämoglobin: hat eine Quartärstruktur. Vier miteinander verbundene Polypeptidketten bilden eine fast regelmäßige Kugelform, wobei jede Kette zwei Ketten berührt.
Hämoglobin ist ein Atmungsfarbstoff, der dem Blut seine rote Farbe verleiht. Hämoglobin besteht aus Protein und Eisenporphyrin und transportiert Sauerstoff von den Atmungsorganen zu Körpergeweben und Kohlendioxid von diesen zu den Atmungsorganen.
Cytochrome- komplexe Proteine (Hämoproteine), die in lebenden Zellen schrittweise Elektronen und/oder Wasserstoff von oxidierbaren organischen Substanzen auf molekularen Sauerstoff übertragen. Dabei entsteht eine energiereiche ATP-Verbindung.
Cobalamine- natürliche biologisch aktive organische Kobaltverbindungen. Die strukturelle Basis von Kobalt ist ein Corrinring, bestehend aus 4 Pyrrolkernen, in denen die Stickstoffatome an das zentrale Kobaltatom gebunden sind.
Physikalisch-chemische Prinzipien des Sauerstofftransports durch Hämoglobin- Atom (Fe (II)) (einer der Bestandteile von Hämoglobin) kann 6 Koordinationsbindungen bilden. Davon dienen vier der Fixierung des Fe(II)-Atoms selbst im Häm, die fünfte Bindung der Bindung des Häms an die Proteinuntereinheit und die sechste Bindung der Bindung des O 2 - bzw. CO 2 -Moleküls.
Metall-Ligand-Homöostase und Ursachen ihrer Verletzung. Mechanismus der toxischen Wirkung von Schwermetallen und Arsen basierend auf der Theorie der harten und weichen Säuren und Basen (HMBA). Thermodynamische Prinzipien der Chelattherapie. Mechanismus der zytotoxischen Wirkung von Platinverbindungen.
Im Körper kommt es kontinuierlich zur Bildung und Zerstörung von Biokomplexen aus Metallkationen und Bioliganden (Porphine, Aminosäuren, Proteine, Polynukleotide), zu denen Spenderatome von Sauerstoff, Stickstoff und Schwefel gehören. Der Austausch mit der Umgebung hält die Konzentrationen dieser Stoffe auf einem konstanten Niveau und liefert Metall Ligand Homöostase. Die Verletzung des bestehenden Gleichgewichts führt zu einer Reihe pathologischer Phänomene - Metallüberschuss- und Metallmangelzustände. Als Beispiel kann eine unvollständige Liste von Krankheiten angeführt werden, die mit Veränderungen im Metall-Liganden-Gleichgewicht nur für ein Ion, das Kupferkation, einhergehen. Ein Mangel an diesem Element im Körper verursacht Menkes-Syndrom, Morfan-Syndrom, Wilson-Konovalov-Krankheit, Leberzirrhose, Emphysem, Aorto- und Arteriopathie, Anämie. Eine übermäßige Aufnahme des Kations kann zu einer Reihe von Erkrankungen verschiedener Organe führen: Rheuma, Bronchialasthma, Nieren- und Leberentzündung, Herzinfarkt usw., Hyperkuperämie genannt. Professionelle Hyperkupreose ist auch bekannt - Kupferfieber.
Die Zirkulation von Schwermetallen erfolgt teilweise in Form von Ionen oder Komplexen mit Aminosäuren, Fettsäuren. Die Hauptrolle beim Transport von Schwermetallen kommt jedoch Proteinen zu, die mit ihnen eine starke Bindung eingehen.
Sie werden auf Zellmembranen fixiert, blockieren die Thiolgruppen von Membranproteinen- 50 % von ihnen sind Protein-Enzyme, die die Stabilität der Protein-Lipid-Komplexe der Zellmembran und ihre Durchlässigkeit stören, was die Freisetzung von Kalium aus der Zelle und das Eindringen von Natrium und Wasser in sie verursacht.
Eine ähnliche Wirkung dieser Gifte, die aktiv an roten Blutkörperchen fixiert werden, führt zu einer Störung der Integrität von Erythrozytenmembranen, einer Hemmung der aeroben Glykolyse und Stoffwechselvorgängen in ihnen im Allgemeinen und einer Akkumulation von hämolytisch aktivem Wasserstoffperoxid aufgrund einer Hemmung der Peroxidase insbesondere, was zur Entwicklung eines der charakteristischen Vergiftungssymptome durch Verbindungen dieser Gruppe führt - zur Hämolyse.
Die Verteilung und Ablagerung von Schwermetallen und Arsen erfolgt in fast allen Organen. Von besonderem Interesse ist die Fähigkeit dieser Substanzen, sich in den Nieren anzusammeln, was durch den reichen Gehalt an Thiolgruppen im Nierengewebe und das Vorhandensein eines Proteins darin erklärt wird - Metallobionin, das eine große Anzahl von Thiolgruppen enthält, die trägt zur langfristigen Ablagerung von Giften bei. Auch das an Thiolgruppen reiche und metallobioninhaltige Lebergewebe zeichnet sich durch eine hohe Akkumulation toxischer Verbindungen dieser Gruppe aus. Die Lagerfrist von beispielsweise Quecksilber kann 2 Monate und mehr betragen.
Die Ausscheidung von Schwermetallen und Arsen erfolgt in unterschiedlichen Anteilen über Nieren, Leber (mit Galle), Magen- und Darmschleimhaut (mit Kot), Schweiß- und Speicheldrüsen, Lunge, was meist mit einer Schädigung des Ausscheidungsapparates einhergeht dieser Organe und äußert sich in den entsprechenden klinischen Symptomen.
Die tödliche Dosis für lösliche Quecksilberverbindungen beträgt 0,5 g, für Calomel 1–2 g, für Kupfersulfat 10 g, für Bleiacetat 50 g, für Bleiweiß 20 g, für Arsen 0,1–0,2 g.
Die Quecksilberkonzentration im Blut beträgt mehr als 10 µg/l (1γ%), im Urin mehr als 100 µg/l (10γ%), die Kupferkonzentration im Blut beträgt mehr als 1600 µg/l (160γ% ) Arsen mehr als 250 µg/l (25γ%) %) im Urin.
Chelat-Therapie ist die Entfernung von toxischen Partikeln
aus dem Körper, basierend auf ihrer Chelatbildung
s-Element-Komplexonate.
Drogen verwendet, um zu entfernen
in den Körper von giftigen aufgenommen
Partikel werden als Entgifter bezeichnet.
Kapitel 17
17.1. Grundlegende Definitionen
In diesem Kapitel werden Sie in eine spezielle Gruppe komplexer Substanzen eingeführt, die als umfassend(oder koordinieren) Verbindungen.
Derzeit ist eine strenge Definition des Begriffs " komplexes Teilchen" Nein. Üblicherweise wird die folgende Definition verwendet.
Beispielsweise ist ein hydratisiertes Kupferion 2 ein komplexes Teilchen, da es tatsächlich in Lösungen und einigen kristallinen Hydraten existiert, es aus Cu 2 -Ionen und H 2 O-Molekülen gebildet wird, Wassermoleküle echte Moleküle sind und Cu 2 -Ionen in Kristallen existieren vieler Kupferverbindungen. Im Gegensatz dazu ist das SO 4 2 -Ion kein komplexes Teilchen, da O 2 -Ionen zwar in Kristallen vorkommen, das S 6 -Ion jedoch nicht in chemischen Systemen existiert.
Beispiele für andere komplexe Teilchen: 2 , 3 , , 2 .
Gleichzeitig werden NH 4 - und H 3 O-Ionen als komplexe Teilchen klassifiziert, obwohl H-Ionen in chemischen Systemen nicht vorkommen.
Manchmal werden komplexe Teilchen als komplexe chemische Teilchen bezeichnet, bei denen alle oder ein Teil der Bindungen gemäß dem Donor-Akzeptor-Mechanismus gebildet werden. Dies trifft auf die meisten Komplexpartikel zu, aber beispielsweise in Kaliumalaun SO 4 wird in Komplexpartikel 3 die Bindung zwischen Al- und O-Atomen tatsächlich nach dem Donor-Akzeptor-Mechanismus gebildet, während es in dem Komplexpartikel nur eine elektrostatische Bindung gibt (Ion-Dipol) Wechselwirkung. Dies wird durch die Existenz eines komplexen Teilchens ähnlicher Struktur in Eisen-Ammonium-Alaun bestätigt, in dem nur eine Ion-Dipol-Wechselwirkung zwischen Wassermolekülen und dem NH 4 -Ion möglich ist.
Durch Ladung können komplexe Teilchen Kationen, Anionen und auch neutrale Moleküle sein. Komplexe Verbindungen, die solche Partikel enthalten, können verschiedenen Klassen von Chemikalien (Säuren, Basen, Salze) angehören. Beispiele: (H 3 O) - Säure, OH - Base, NH 4 Cl und K 3 - Salze.
Typischerweise ist der Komplexbildner ein Atom eines Elements, das ein Metall bildet, aber es kann auch ein Atom von Sauerstoff, Stickstoff, Schwefel, Iod und anderen Elementen sein, die Nichtmetalle bilden. Der Oxidationszustand des Komplexbildners kann positiv, negativ oder Null sein; wenn aus einfacheren Substanzen eine komplexe Verbindung gebildet wird, ändert sie sich nicht.
Liganden können Partikel sein, die vor der Bildung einer Komplexverbindung Moleküle (H 2 O, CO, NH 3 usw.), Anionen (OH, Cl, PO 4 3 usw.) sowie ein Wasserstoffkation waren . Unterscheiden nicht identifiziert oder einzähnige Liganden (mit dem Zentralatom über eines seiner Atome verbunden, also über eine -Bindung), zweizähnig(durch zwei ihrer Atome, also durch zwei -Bindungen, mit dem Zentralatom verbunden), dreizähnig usw.
Wenn die Liganden unidentisch sind, dann ist die Koordinationszahl gleich der Anzahl solcher Liganden.
Die cn hängt von der elektronischen Struktur des Zentralatoms, seinem Oxidationsgrad, der Größe des Zentralatoms und der Liganden, den Bedingungen für die Bildung der Komplexverbindung, der Temperatur und anderen Faktoren ab. CN kann Werte von 2 bis 12 annehmen. Meistens ist es gleich sechs, etwas seltener - vier.
Es gibt auch komplexe Teilchen mit mehreren Zentralatomen.
Es werden zwei Arten von Strukturformeln komplexer Teilchen verwendet: Angabe der formalen Ladung des Zentralatoms und der Liganden oder Angabe der formalen Ladung des gesamten komplexen Teilchens. Beispiele:

Um die Form eines komplexen Teilchens zu charakterisieren, wird die Idee eines Koordinationspolyeders (Polyeders) verwendet.

Koordinationspolyeder umfassen auch ein Quadrat (KN = 4), ein Dreieck (KN = 3) und eine Hantel (KN = 2), obwohl diese Figuren keine Polyeder sind. Beispiele für Koordinationspolyeder und entsprechend geformte Komplexteilchen für die gängigsten CN-Werte sind in Abb. eines.
17.2. Klassifizierung komplexer Verbindungen
Wie chemische komplexe Verbindungen in ionische Verbindungen unterteilt werden (sie werden manchmal als ionogen) und molekular ( nichtionisch) Verbindungen. Ionische Komplexverbindungen enthalten geladene Komplexteilchen – Ionen – und sind Säuren, Basen oder Salze (siehe § 1). Molekulare Komplexverbindungen bestehen aus ungeladenen Komplexteilchen (Molekülen), zB: oder - sie lassen sich nur schwer einer Hauptklasse von Chemikalien zuordnen.
Die komplexen Teilchen, aus denen komplexe Verbindungen bestehen, sind sehr unterschiedlich. Daher werden mehrere Klassifizierungsmerkmale für ihre Klassifizierung verwendet: die Anzahl der Zentralatome, die Art des Liganden, die Koordinationszahl und andere.
Nach der Anzahl der Zentralatome Komplexe Teilchen werden unterteilt in Einzelprozessor und mehrkernig. Die Zentralatome mehrkerniger Komplexteilchen können entweder direkt oder über Liganden miteinander verknüpft sein. In beiden Fällen bilden die Zentralatome mit Liganden eine einzige innere Sphäre der Komplexverbindung:
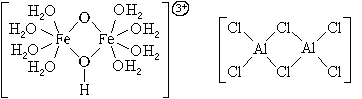

Komplexe Partikel werden nach Art der Liganden unterteilt
1) Aquakomplexe, also komplexe Teilchen, in denen Wassermoleküle als Liganden vorhanden sind. Kationische Aquakomplexe m sind mehr oder weniger stabil, anionische Aquakomplexe sind instabil. Alle kristallinen Hydrate sind Verbindungen, die Aquakomplexe enthalten, zum Beispiel:
Mg(ClO4) 2. 6H 2 O ist eigentlich (ClO 4 ) 2 ;
BeSO4. 4H 2 O ist eigentlich SO 4 ;
Zn(BrO 3 ) 2 . 6H 2 O ist eigentlich (BrO 3 ) 2 ;
CuSO4. 5H 2 O ist eigentlich SO 4 . H2O.
2) Hydroxokomplexe, dh Komplexpartikel, in denen Hydroxylgruppen als Liganden vorhanden sind, die vor Eintritt in das Komplexpartikel Hydroxidionen waren, z. B.: 2 , 3 , .
Hydroxo-Komplexe werden aus Aqua-Komplexen gebildet, die die Eigenschaften von kationischen Säuren aufweisen:
2 + 4OH = 2 + 4H 2 O
3) Ammoniak, dh komplexe Partikel, in denen NH 3 -Gruppen als Liganden vorhanden sind (vor der Bildung eines komplexen Partikels - Ammoniakmoleküle), zum Beispiel: 2 , , 3 .
Ammoniak kann auch aus Wasserkomplexen gewonnen werden, zum Beispiel:
2 + 4NH 3 \u003d 2 + 4 H 2 O
Die Farbe der Lösung ändert sich dabei von Blau nach Ultramarin.
4) Säurekomplexe, dh komplexe Partikel, in denen saure Reste sowohl sauerstofffreier als auch sauerstoffhaltiger Säuren als Liganden vorhanden sind (vor der Bildung eines komplexen Partikels - Anionen, zum Beispiel: Cl, Br, I, CN, S 2, NO 2, S 2 O 3 2 , CO 3 2 , C 2 O 4 2 usw.).
Beispiele für die Bildung von Säurekomplexen:
Hg2 + 4I = 2
AgBr + 2S 2 O 3 2 = 3 + Br
Die letztere Reaktion wird in der Fotografie verwendet, um nicht umgesetztes Silberbromid aus fotografischen Materialien zu entfernen.
(Bei der Entwicklung von Fotofilmen und Fotopapieren wird der unbelichtete Teil des in der Fotoemulsion enthaltenen Silberbromids nicht vom Entwickler wiederhergestellt. Um es zu entfernen, wird diese Reaktion verwendet (der Vorgang wird "Fixieren" genannt, da das nicht entfernte Silberbromid zersetzt sich allmählich im Licht und zerstört das Bild)
5) Komplexe, in denen Wasserstoffatome Liganden sind, werden in zwei völlig unterschiedliche Gruppen eingeteilt: Hydrid Komplexe und Komplexe, die in der Zusammensetzung enthalten sind Onium Verbindungen.
Bei der Bildung von Hydridkomplexen - , , - ist das Zentralatom ein Elektronenakzeptor und das Hydridion ein Donor. Die Oxidationsstufe der Wasserstoffatome in diesen Komplexen ist –1.
In Oniumkomplexen ist das Zentralatom ein Elektronendonor und der Akzeptor ein Wasserstoffatom in der Oxidationsstufe +1. Beispiele: H 3 O oder - Oxoniumion, NH 4 oder - Ammoniumion. Darüber hinaus gibt es substituierte Derivate solcher Ionen: - Tetramethylammonium-Ion, - Tetraphenylarsonium-Ion, - Diethyloxonium-Ion usw.
6) Carbonyl Komplexe - Komplexe, in denen CO-Gruppen als Liganden vorhanden sind (vor der Komplexbildung - Kohlenmonoxidmoleküle), zum Beispiel: usw.
7) Anion Halogenid Komplexe sind Komplexe vom Typ .
Auch andere Klassen von Komplexpartikeln werden nach der Art der Liganden unterschieden. Darüber hinaus gibt es komplexe Partikel mit Liganden verschiedener Art; Das einfachste Beispiel ist Aqua Hydroxocomplex.
17.3. Grundlagen der Nomenklatur komplexer Verbindungen
Die Formel einer komplexen Verbindung wird auf die gleiche Weise zusammengestellt wie die Formel einer ionischen Substanz: An erster Stelle steht die Formel des Kations und an zweiter Stelle die des Anions.
Die Formel eines Komplexteilchens wird in eckige Klammern in folgender Reihenfolge geschrieben: Zuerst wird das Symbol des komplexbildenden Elements gesetzt, dann die Formeln der Liganden, die vor der Komplexbildung Kationen waren, dann die Formeln der Liganden, die es waren neutrale Moleküle vor der Komplexbildung und danach die Formeln der Liganden, ehemals vor der Komplexbildung durch Anionen.
Der Name einer komplexen Verbindung wird genauso aufgebaut wie der Name eines beliebigen Salzes oder einer Base (komplexe Säuren werden als Wasserstoff- oder Oxoniumsalze bezeichnet). Der Name der Verbindung umfasst den Namen des Kations und den Namen des Anions.
Der Name des Komplexpartikels enthält den Namen des Komplexbildners und die Namen der Liganden (der Name wird gemäß der Formel geschrieben, jedoch von rechts nach links. Für Komplexbildner in Kationen werden russische Elementnamen verwendet, und in Anionen, lateinische.
Namen der häufigsten Liganden:
| H 2 O - Wasser | Cl - Chlor | SO 4 2 - Sulfat | OH - hydroxo |
| CO - Carbonyl | Br - Brom | CO 3 2 - Karbonat | H - Hydrido |
| NH 3 - Ammin | NO 2 - Nitro | CN - Cyano | NEIN - Nitroso |
| NO - Nitrosyl | O 2 - Oxo | NCS - Thiocyanato | H + I - hydro |
Beispiele für Namen komplexer Kationen:
Beispiele für Namen komplexer Anionen:
2 - Tetrahydroxozinkation
3 – Di(thiosulfato)argentat(I)-ion
3 – Hexacyanochromat(III)-Ion
– Tetrahydroxodiquaaluminat-Ion
– Tetranitrodiamincobaltat(III)-ion
3 – Pentacyanoaquaferrat(II)-ion
Beispiele für die Namen neutraler Komplexteilchen:
Ausführlichere Nomenklaturregeln sind in Nachschlagewerken und speziellen Handbüchern angegeben.
17.4. Chemische Bindung in komplexen Verbindungen und ihre Struktur
Bei kristallinen Komplexverbindungen mit geladenen Komplexen ist die Bindung zwischen dem Komplex und den Ionen der äußeren Sphäre ionisch, während die Bindungen zwischen den restlichen Teilchen der äußeren Sphäre intermolekular (einschließlich Wasserstoffbrückenbindungen) sind. Bei molekularen Komplexverbindungen ist die Bindung zwischen den Komplexen intermolekular.
In den meisten komplexen Partikeln sind die Bindungen zwischen dem Zentralatom und den Liganden kovalent. Alle oder ein Teil von ihnen werden nach dem Donor-Akzeptor-Mechanismus gebildet (als Ergebnis mit einer Änderung der formalen Ladungen). In den am wenigsten stabilen Komplexen (z. B. in den Aquakomplexen von Alkali- und Erdalkalielementen sowie Ammonium) werden Liganden durch elektrostatische Anziehung gehalten. Die Bindung in komplexen Partikeln wird oft als Donor-Akzeptor- oder Koordinationsbindung bezeichnet.
Betrachten wir seine Entstehung am Beispiel der Eisen(II)-Aquakation. Dieses Ion wird durch die Reaktion gebildet:
FeCl2cr + 6H20 = 2 + 2Cl
Die elektronische Formel des Eisenatoms ist 1 s 2 2s 2 2p 6 3s 2 3p 6 4s 2 3d 6. Lassen Sie uns ein Schema der Valenz-Unterebenen dieses Atoms erstellen:
Wenn ein doppelt geladenes Ion entsteht, verliert das Eisenatom zwei 4 s-Elektron:
Das Eisenion nimmt sechs Elektronenpaare von Sauerstoffatomen von sechs Wassermolekülen in freie Valenzorbitale auf:

Es entsteht ein komplexes Kation, dessen chemische Struktur durch eine der folgenden Formeln ausgedrückt werden kann:

Die räumliche Struktur dieses Teilchens wird durch eine der räumlichen Formeln ausgedrückt:

Die Form des Koordinationspolyeders ist ein Oktaeder. Alle Fe-O-Bindungen sind gleich. Soll sp 3 d 2 - Hybridisierung des Eisenatoms AO. Die magnetischen Eigenschaften des Komplexes weisen auf das Vorhandensein ungepaarter Elektronen hin.
Löst man FeCl 2 in einer Cyanidionen enthaltenden Lösung, so läuft die Reaktion ab
FeCl2cr + 6CN = 4 + 2Cl.
Derselbe Komplex wird auch durch Zugabe einer Lösung von Kaliumcyanid KCN zu einer FeCl 2 -Lösung erhalten:
2 + 6CN \u003d 4 + 6H 2 O.
Dies deutet darauf hin, dass der Cyanidkomplex stärker als der Aquakomplex ist. Darüber hinaus weisen die magnetischen Eigenschaften des Cyanidkomplexes auf das Fehlen ungepaarter Elektronen vom Eisenatom hin. All dies ist auf eine etwas andere elektronische Struktur dieses Komplexes zurückzuführen:

Die "stärkeren" CN-Liganden bilden stärkere Bindungen mit dem Eisenatom, der Energiegewinn reicht aus, um die Hundsche Regel zu "brechen" und 3 freizusetzen d-Orbitale für freie Ligandenpaare. Die räumliche Struktur des Cyanidkomplexes ist die gleiche wie die des Aquakomplexes, aber die Art der Hybridisierung ist anders - d 2 sp 3 .
Die "Stärke" des Liganden hängt in erster Linie von der Elektronendichte der Wolke des freien Elektronenpaars ab, das heißt, sie nimmt mit abnehmender Atomgröße zu, mit abnehmender Hauptquantenzahl hängt davon ab Art der EO-Hybridisierung und einigen anderen Faktoren. Die wichtigsten Liganden können aneinandergereiht werden, um ihre "Stärke" zu erhöhen (eine Art "Aktivitätsreihe" von Liganden), diese Reihe wird genannt spektrochemische Reihe von Liganden:
| ICH; Br; : SCN, Cl, F, OH, H 2 O; : NCS, NH3; SO 3 S : 2 ; : CN, CO |
Für die Komplexe 3 und 3 sehen die Bildungsschemata wie folgt aus:
|
|


Für Komplexe mit CN = 4 sind zwei Strukturen möglich: ein Tetraeder (in dem Fall sp 3-Hybridisierung), zum Beispiel 2 , und ein flaches Quadrat (im Fall von dsp 2-Hybridisierung), zum Beispiel 2 .
17.5. Chemische Eigenschaften komplexer Verbindungen
Für komplexe Verbindungen sind zunächst dieselben Eigenschaften charakteristisch wie für gewöhnliche Verbindungen derselben Klassen (Salze, Säuren, Basen).
Wenn die Verbindung eine Säure ist, dann ist es eine starke Säure, wenn es eine Base ist, dann ist die Base stark. Diese Eigenschaften von Komplexverbindungen werden nur durch die Anwesenheit von H 3 O- oder OH-Ionen bestimmt. Außerdem gehen komplexe Säuren, Basen und Salze die üblichen Austauschreaktionen ein, zum Beispiel:
SO 4 + BaCl 2 \u003d BaSO 4 + Cl 2
FeCl 3 + K 4 = Fe 4 3 + 3KCl
Die letzte dieser Reaktionen wird als qualitative Reaktion für Fe 3 -Ionen verwendet. Die resultierende ultramarin-unlösliche Substanz wird "Preußischblau" genannt [der systematische Name ist Eisen(III)-Kaliumhexacyanoferrat(II)].
Außerdem kann das komplexe Teilchen selbst in die Reaktion eintreten, und je aktiver, desto weniger stabil ist es. Normalerweise sind dies Ligandensubstitutionsreaktionen, die in Lösung stattfinden, zum Beispiel:
2 + 4NH 3 \u003d 2 + 4H 2 O,
sowie Säure-Base-Reaktionen wie z
2 + 2H 3 O = + 2H 2 O
2 + 2OH = + 2H 2 O
In diesen Reaktionen gebildet, wird es nach Isolierung und Trocknung zu Zinkhydroxid:
Zn(OH) 2 + 2H 2 O
Die letzte Reaktion ist das einfachste Beispiel für die Zersetzung einer komplexen Verbindung. In diesem Fall läuft es bei Raumtemperatur. Andere komplexe Verbindungen zersetzen sich beim Erhitzen, zum Beispiel:
SO4. H 2 O \u003d CuSO 4 + 4NH 3 + H 2 O (über 300 ° C)
4K 3 \u003d 12KNO 2 + 4CoO + 4NO + 8NO 2 (über 200 ° C)
K 2 \u003d K 2 ZnO 2 + 2 H 2 O (über 100 ° C)
Um die Möglichkeit einer Ligandensubstitutionsreaktion abzuschätzen, kann eine spektrochemische Reihe verwendet werden, die sich an der Tatsache orientiert, dass stärkere Liganden schwächere aus der inneren Sphäre verdrängen.
17.6. Isomerie komplexer Verbindungen
Die Isomerie komplexer Verbindungen ist verwandt
1) mit möglicher unterschiedlicher Anordnung von Liganden und Partikeln der äußeren Sphäre,
2) mit einer anderen Struktur des komplexesten Teilchens.
Zur ersten Gruppe gehören hydratisiert(Im Algemeinen lösen) und Ionisation Isomerie, zum zweiten - räumlich und optisch.
Hydratisomerie ist mit der Möglichkeit einer unterschiedlichen Verteilung von Wassermolekülen in den äußeren und inneren Sphären der Komplexverbindung verbunden, zum Beispiel: (rotbraune Farbe) und Br 2 (blaue Farbe).
Ionisationsisomerie ist mit der Möglichkeit einer unterschiedlichen Verteilung von Ionen in den äußeren und inneren Sphären verbunden, zum Beispiel: SO 4 (lila) und Br (rot). Die erste dieser Verbindungen bildet einen Niederschlag, der mit einer Lösung von Bariumchlorid reagiert, und die zweite - mit einer Lösung von Silbernitrat.
Räumliche (geometrische) Isomerie, auch cis-trans-Isomerie genannt, ist charakteristisch für quadratische und oktaedrische Komplexe (für tetraedrische Komplexe ist dies unmöglich). Beispiel: cis-trans-Quadratkomplex-Isomerie

Die optische (Spiegel-)Isomerie unterscheidet sich im Wesentlichen nicht von der optischen Isomerie in der organischen Chemie und ist charakteristisch für tetraedrische und oktaedrische Komplexe (unmöglich für quadratische).
komplexe Verbindungen. Ihre Struktur basiert auf der Koordinationstheorie von A. Werner. Komplexes Ion, seine Ladung. Kationische, anionische, neutrale Komplexe. Nomenklatur, Beispiele.



Ligandensubstitutionsreaktionen. Komplexe Ioneninstabilitätskonstante, Stabilitätskonstante.

 Die Instabilität ist das Verhältnis der Produkte der Konzentration von zerfallenen Ionen zu der nicht zerfallenen Menge.
Die Instabilität ist das Verhältnis der Produkte der Konzentration von zerfallenen Ionen zu der nicht zerfallenen Menge.
K set \u003d 1 / K nest (reziprok)
 Sekundäre Dissoziation - die Auflösung der inneren Sphäre des Komplexes in ihre Bestandteile.
Sekundäre Dissoziation - die Auflösung der inneren Sphäre des Komplexes in ihre Bestandteile.
43. Konkurrenz um einen Liganden oder um einen Komplexbildner: isolierte und kombinierte Gleichgewichte der Ligandensubstitution. Allgemeine Konstante des kombinierten Gleichgewichts der Ligandensubstitution.
Als Ergebnis der Konkurrenz zerstört das Proton einen ausreichend starken Komplex und bildet eine schwach dissoziierende Substanz - Wasser.
Cl + NiS0 4 +4NH 3 ^ S0 4 + AgCl I
Dies ist bereits ein Beispiel für Ligandenkonkurrenz um einen Komplexbildner mit der Bildung eines stabileren Komplexes (K H + \u003d 9,3-1 (G 8; K H [M (W 3) 6 ] 2+ \u003d 1,9-10 - 9) und eine schwerlösliche Verbindung AgCl - K s \u003d 1,8 · 10 "10
Vorstellungen über die Struktur von Metalloenzymen und anderen Biokomplexverbindungen (Hämoglobin, Cytochrome, Cobalamine). Physikalisch-chemische Prinzipien des Sauerstofftransports durch Hämoglobin



Cobalamine. Vitamin B12 eine Gruppe kobalthaltiger biologisch aktiver Substanzen, die als Cobalamine bezeichnet werden. Das sind sie tatsächlich Cyanocobalamin, Hydroxycobalamin und zwei coenzymatische Formen von Vitamin B 12: Methylcobalamin und 5-Desoxyadenosylcobalamin.
Manchmal wird Vitamin B 12 im engeren Sinne als Cyanocobalamin bezeichnet, da in dieser Form die Hauptmenge an Vitamin B 12 in den menschlichen Körper gelangt, ohne dabei aus den Augen zu verlieren, dass es nicht gleichbedeutend mit B 12 und mehreren ist andere Verbindungen haben auch B 12 -Vitamin-Aktivität. Vitamin B 12 wird auch Castles extrinsischer Faktor genannt.
B 12 hat im Vergleich zu anderen Vitaminen die komplexeste chemische Struktur, deren Grundlage der Corrin-Ring ist. Corrin ähnelt in vielerlei Hinsicht Porphyrin (eine komplexe chemische Struktur, die Teil von Häm, Chlorophyll und Cytochromen ist), unterscheidet sich jedoch von Porphyrin dadurch, dass zwei Pyrrolringe in der Zusammensetzung von Corrin direkt miteinander verbunden sind und nicht durch ein Methylen Brücke. Das Cobalt-Ion befindet sich im Zentrum der Corrin-Struktur. Cobalt bildet vier Koordinationsbindungen mit Stickstoffatomen. Eine weitere Koordinationsbindung verbindet Cobalt mit dem Nukleotid Dimethylbenzimidazol. Die letzte, sechste Koordinationsbindung von Cobalt bleibt frei: Durch diese Bindung wird die Cyanogruppe, Hydroxylgruppe, Methyl- oder 5 "-Desoxyadenosylrest hinzugefügt, um jeweils vier Varianten von Vitamin B 12 zu bilden. Die Kohlenstoff-kovalente kovalente Bindung In der Struktur von Cyanocobalamin ist das einzige in der belebten Natur bekannte Beispiel für eine kovalente Bindung Übergangsmetall-Kohlenstoff.
