Ligand substitution reactions. Complex ion instability constant, stability constant
The elementary stages of organic reactions catalyzed by acids, bases, nucleophilic catalysts, metal complexes, solid metals and their compounds in gas-phase or liquid-phase heterogeneous and homogeneous processes are reactions of the formation and transformation of various organic and organometallic intermediates, as well as metal complexes. Organic intermediates include carbenium ions R + , carbonium RH 2 + , carbo-anions R-, anion- and cation radicals, radicals and biradicals R , R:, as well as molecular complexes of organic donor and acceptor molecules (D A), which are called also complexes with charge transfer. In homogeneous and heterogeneous catalysis by metal complexes (metal complex catalysis) of organic reactions, the intermediates are complex (coordination) compounds with organic and inorganic ligands, organometallic compounds with an M–C bond, which in most cases are coordination compounds. A similar situation takes place in the case of “two-dimensional” chemistry on the surface of solid metal catalysts. Let us consider the main types of reactions of metal complexes and organometallic compounds.
Elementary steps involving metal complexesThe reactions of metal complexes can be divided into three groups:
a) electron transfer reactions;
b) ligand substitution reactions;
c) reactions of coordinated ligands.
Electron transfer reactions
Two mechanisms are implemented in electron transfer reactions - the outer sphere mechanism (without changes in the coordination spheres of the donor and acceptor) and the bridging (inner sphere) mechanism, leading to changes in the coordination sphere of the metal.
Let us consider the outer sphere mechanism using the example of octahedral complexes of transition metals. In the case of symmetrical reactions ( G 0 = 0)
the rate constants vary in a very wide range of values - from 10-12 to 10 5 l mol-1 sec-1, depending on the electronic configuration of the ion and the degree of its ᴨȇrestructuring during the process. In these reactions, the principle of least movement is very clearly manifested - the least change in the valence shell of the participants in the reaction.
In the reaction of electron transfer (1) (Co * is an isotope of the Co atom)
(symmetrical reaction), Co 2+ (d 7) ᴨȇ goes into Co 3+ (d 6). The electronic configuration (valence shell) does not change during this transition
6 electrons on the triply degenerate bonding level remain unchanged (), and from the antibonding e g level, one electron is removed.
Second order rate constant for reaction (1) k 1 \u003d 1.1 lmol-1 sec-1. Since Phen (phenanthroline) is a strong ligand, the maximum number of 7 d-electrons are paired (spin-paired state). In the case of a weak NH3 ligand, the situation changes dramatically. Co(NH 3) n 2+ (n = 4, 5, 6) is in the spin-unpaired (high-spin) state.
The stronger Co(NH 3) 6 3+ complex (~10 30 times stronger than Co(NH 3) 6 2+) is in a spin-paired state, as is the complex with Phen. In this regard, in the process of electron transfer, the valence shell must be strongly rebuilt and, as a result, k\u003d 10-9 lmol-1 sec-1. The degree of conversion of Co 2+ to Co 3+, equal to 50%, is achieved in the case of the Phen ligand in 1 second, and in the case of NH 3 ~ in 30 years. Obviously, a stage with such a rate (formally elementary) can be excluded from the set of elementary stages in the analysis of reaction mechanisms.
Value G for the electron transfer reaction during the formation of a collision complex, according to the Marcus theory, it includes two components and
The first term is the energy of the reorganization of the M-L bonds within the complex (the length and strength of the bond with a change in the valence state). The value includes the energy of the rearrangement of the outer solvate shell in the process of changing the M-L coordinates and the charge of the complex. The smaller the change in the electronic environment and the smaller the change in the length M-L, the lower, the larger the ligands, the smaller and, as a result, the higher the rate of electron transfer. The value for the general case can be calculated using the Marcus equation
where. At = 0 .
In the case of an intrasphere mechanism, the process of electron transfer is facilitated, since one of the ligands of the first complex forms a bridge complex with the second complex, displacing one of the ligands from it
The rate constants of such a process are 8 orders of magnitude higher than the constants for the reduction of Cr(NH 3) 6 3+ . In such reactions, the reducing agent must be a labile complex, and the ligand in the oxidizing agent must be able to form bridges (Cl-, Br-, I-, N 3 -, NCS-, bipy).
Ligand substitution reactionsOne of the most important stages in metal complex catalysis, the interaction of the substrate Y with the complex, proceeds by three mechanisms:
a) Replacement of the ligand with a solvent. Usually such a stage is depicted as the dissociation of the complex
The essence of the process in most cases is the replacement of the ligand L by the solvent S, which is then easily replaced by the substrate molecule Y
b) Attachment of a new ligand along a free coordinate with the formation of an associate, followed by dissociation of the substituted ligand
c) Synchronous substitution (type S N 2) without the formation of an intermediate
In the case of Pt(II) complexes, the reaction rate is very often described by the two-way equation
where k S and k Y are the rate constants of the processes occurring in reactions (5) (with solvent) and (6) with ligand Y. For example,
The last stage of the second route is the sum of three fast elementary steps - cleavage of Cl-, addition of Y, and elimination of the H 2 O molecule.
In planar square complexes of ᴨȇtransition metals, a trans-effect is observed, formulated by I.I. For Pt(II) complexes, the trans effect increases in the series of ligands:
H2O~NH3< Cl- ~ Br- < I- ~ NO 2 - ~ C 6 H 5 - < CH 3 - <
< PR 3 ~ AsR 3 ~ H- < олефин ~ CO ~ CN-.
The presence of the kinetic trans effect and thermodynamic trans effect explains the possibility of synthesizing inert isomeric complexes Pt(NH 3) 2 Cl 2:
Reactions of coordinated ligands§ Reactions of electrophilic substitution (S E) of hydrogen by metal in the coordination sphere of the metal and their reverse processes
SH - H 2 O, ROH, RNH 2, RSH, ArH, RCCH.
Even H 2 and CH 4 molecules are involved in reactions of this type
§ Insertion reactions L over M-X bond
In the case of X = R (an organometallic complex), metal-coordinated molecules are also introduced at the M-R bond (L - CO, RNC, C 2 H 2 , C 2 H 4 , N 2 , CO 2 , O 2 , etc.). Insertion reactions are the result of an intramolecular attack of the nucleophile X on a molecule coordinated by - or -type. Reverse reactions - reactions - and -elimination
§ Reactions of oxidative addition and reductive elimination
M 2 (C 2 H 2) M 2 4+ (C 2 H 2) 4-
Apparently, in these reactions there is always a preliminary coordination of the attached molecule, but this is not always possible to fix. In this regard, the presence of a free site in the coordination sphere or a site associated with a solvent, which is easily replaced by a substrate, is an important factor affecting the reactivity of metal complexes. For example, bis-allyl complexes of Ni are good precursors of catalytically active species, since due to the easy reductive elimination of bis-allyl, a complex with a solvent, the so-called. bare nickel. The role of free seats is illustrated by the following example:
§ Reactions of nucleophilic and electrophilic addition to - and - metal complexes
Reactions of organometallic compoundsAs intermediates in catalytic reactions, there are both classical organometallic compounds having M–C, M=C, and MC bonds, as well as nonclassical compounds, in which the organic ligand is coordinated according to the 2, 3, 4, 5, and 6 type, or is an element of electron-deficient structures - bridging CH 3 and C 6 H 6 groups, non-classical carbides (Rh 6 C (CO) 16, C (AuL) 5 +, C (AuL) 6 2+, etc.).
Among the specific mechanisms for classical -organometallic compounds, we note several mechanisms. Thus, 5 mechanisms of electrophilic substitution of a metal atom at the M-C bond have been established.
electrophilic substitution with nucleophilic assistance
AdE Addition-elimination
AdE(C) Attachment to the C atom in sp 2 hybridization
AdE(M) Oxidative addition to metal
Nucleophilic substitution at the carbon atom in the reactions of demetallation of organometallic compounds occurs as a redox process:
It is possible that an oxidizing agent may be involved in this step.
CuCl 2 , p-benzoquinone, NO 3 - and other compounds can serve as such an oxidizing agent. Here are two more elementary stages characteristic of RMX:
M-C bond hydrogenolysis
and homolysis of the M-C bond
An important rule relating to all reactions of complex and organometallic compounds and related to the principle of least motion is Tolman's 16-18 electron shell rule (Section 2).
Coordination and organometallic compoundson a surfaceAccording to modern concepts, complexes and organometallic compounds are formed on the surface of metals, similar to compounds in solutions. For surface chemistry, the participation of several surface atoms in the formation of such compounds and, of course, the absence of charged particles is essential.
Surface groups can be any atoms (H, O, N, C), groups of atoms (OH, OR, NH, NH 2 , CH, CH 2 , CH 3 , R), coordinated molecules CO, N 2 , CO 2 , C 2 H 4 , C 6 H 6 . For example, during the adsorption of CO on the surface of a metal, the following structures were found:
The C 2 H 4 molecule on the metal surface forms α-complexes with one center and di-linked ethylene bridges M-CH 2 CH 2 -M, i.e. Essentially, metallocycles
On the surface of Rh, for example, during the adsorption of ethylene, the following processes of ethylene transformation occur as the temperature rises:
The reactions of surface intermediates include the stages of oxidative addition, reductive elimination, insertion, - and -elimination, hydrogenolysis of M-C and C-C bonds, and other reactions of the organometallic type, but without the appearance of free ions. The tables list the mechanisms and intermediates of surface transformations of hydrocarbons on metals.
Table 3.1. Catalytic reactions involving C-C bond breaking.
Designations:
Alkyl, metallacycline;
Carben, allyl;
Carbine, vinyl.
Table 3.2. Catalytic reactions involving the formation of a C-C bond.
Designations: see table. 3.1.
The formation of all the above organometallic compounds on the surface of metals was confirmed by physical methods.
Questions for self-control
1) How does the rule of least change in the valence shell of a metal during ES manifest itself in electron transfer reactions?
2) Why do coordination vacancies contribute to effective interaction with the substrate?
3) List the main types of reactions of coordinated ligands.
4) Give the mechanisms of electrophilic substitution in the reactions of organometallic compounds with HX.
5) Give examples of surface organometallic compounds.
6) Give examples of the participation of metal carbene surface complexes in the transformations of hydrocarbons.
Literature for in-depth study
1. Temkin O.N., Kinetics of catalytic reactions in solutions of metal complexes, M., MITHT, 1980, Part III.
2. Kollman J., Higedas L., Norton J., Finke R., Organometallic Chemistry of Transition Metals, M., Mir, 1989, vol. I, vol. II.
3. Moiseev I.I., -Complexes in the oxidation of olefins, M., Nauka, 1970.
4. O. N. Temkin, G. K. Shestakov, Yu. A. Treger, Acetylene: Chemistry. Reaction mechanisms. Technology. M., Chemistry, 1991, 416 p., section 1.
5. Henrici-Olive G., Olive S., Coordination and catalysis, M., Mir, 1980, 421 p.
6. O. V. Krylov and V. A. Matyshak, Intermediate Compounds in Heterogeneous Catalysis, Moscow, Nauka, 1996.
7. Zaera F., An Organometallic Guide to the Chemistry of Hydrocarbon Moities on Transition Metal Surfaces., Chem. Rev., 1995, 95, 2651-2693.
8. Bent B.E., Mimicking Aspects of Heterogeneous Catalysis: Generating, Isolating, and Reacting Proposed Surface Intermediates on Single Crystals in Vacuum, Chem. Rev., 1996, 96, 1361-1390.
Reactions of coordination compounds always occur in the coordination sphere of the metal with the ligands bound in it. Therefore, it is obvious that in order for something to happen at all, ligands must be able to fall into this sphere. This can happen in two ways:
- the coordinatively unsaturated complex binds the new ligand
- in an already completed coordination sphere, one ligand changes to another.
We already got acquainted with the first method when we discussed coordination unsaturation and the 18-electron rule. Let's do the second one here.
Ligands of any type can be substituted in any combination
But usually there is an unspoken rule - the number of occupied coordination places does not change. In other words, substitution does not change the electron count. The substitution of a ligand of one type for another is quite possible and often occurs in reality. Let us only pay attention to the correct handling of charges when the L-ligand changes to the X-ligand and vice versa. If we forget about this, then the degree of oxidation of the metal will change, and the replacement of ligands is not a redox process (if you find or come up with a nasty example, let me know - the offset will be automatic immediately if I cannot prove that you were mistaken, why even in In this case, I guarantee a positive contribution to karma).
Substitution involving hapto ligands
With more complex ligands, there is no more difficulty - you just need to remember a fairly obvious rule: the number of ligand sites (that is, the total number of ligands or ligand centers of X- or L-types) is conserved. This follows directly from the conservation of the electron count. Here are some self-evident examples.

Let's look at the last example. The starting reagent for this reaction is iron dichloride FeCl 2 . Until recently, we would have said: “It's just salt, what does the coordination chemistry have to do with it?”. But we will no longer allow ourselves such ignorance. In the chemistry of transition metals, there are no “simply salts”, any derivatives are coordination compounds, to which all the arguments about electron counting, d-configuration, coordination saturation, etc. are applicable. Iron dichloride, as we are used to writing it, would be an MX 2 type Fe(2+) complex with a d 6 configuration and 10 electrons. Not enough! Fine? After all, we have already figured out that ligands are implicit. To make a reaction, we need a solvent, and for such reactions it is most likely THF. The dissolution of the crystalline iron salt in THF occurs precisely because the donor solvent occupies free places, and the energy of this process compensates for the destruction of the crystal lattice. We would not be able to dissolve this "salt" in a solvent that does not provide metal solvation services due to Lewis basicity. In this case, and in a million others like it, solvation is just a coordination interaction. Let us write, just for definiteness, the result of solvation in the form of a FeX 2 L 4 complex, in which two chlorine ions remain in the coordination sphere in the form of two X-ligands, although most likely they are also displaced by donor solvent molecules with the formation of a charged complex FeL 6 2+. In this case, it's not that important. And so, and so we can safely assume that we have an 18-electron complex on the left and on the right.
Substitution, addition and dissociation of ligands are closely and inextricably linked
If we remember organic chemistry, then there were two substitution mechanisms at a saturated carbon atom - SN1 and SN2. In the first, the substitution occurred in two stages: the old substituent first left, leaving a vacant orbital on the carbon atom, which was followed by a new substituent with a pair of electrons. The second mechanism assumed that the departure and arrival are carried out simultaneously, in concert, and the process was one-stage.
In the chemistry of coordination compounds, it is quite possible to imagine something similar. But a third possibility appears, which the saturated carbon atom did not have - first we attach a new ligand, then we unhook the old one. It immediately becomes clear that this third option is hardly possible if the complex already has 18 electrons and is coordinatively saturated. But it is quite possible if the number of electrons is 16 or less, that is, the complex is unsaturated. Let us immediately recall an obvious analogy from organic chemistry - nucleophilic substitution at an unsaturated carbon atom (in an aromatic ring or carbonyl carbon) also goes first as the addition of a new nucleophile, and then the elimination of the old one.
So, if we have 18 electrons, then the substitution goes like a split-off-attachment (fans of “smart” words use the term dissociative-associative or simply dissociative mechanism). Another way would require expanding the coordination sphere to a count of 20 electrons. This is not absolutely impossible, and such options are sometimes even considered, but it is definitely very disadvantageous and every time if such a path is suspected, very weighty evidence is required. In most of these stories, the researchers eventually came to the conclusion that they overlooked or did not take into account something, and the associative mechanism was rejected. So, if the original complex has 18 electrons, then first one ligand must leave, then a new one should come in its place, for example:

If we want to introduce a hapto ligand that occupies several positions into the coordination sphere, we must first release them all. As a rule, this occurs only under sufficiently severe conditions, for example, in order to replace three carbonyls with η 6 -benzene in chromium carbonyl, the mixture is heated under pressure for many hours, from time to time bleed off the released carbon monoxide. Although the scheme depicts the dissociation of three ligands with the formation of a very unsaturated complex with 12 electrons, in reality the reaction most likely occurs in stages, leaving one carbonyl, and benzene enters the sphere, gradually increasing the hapticity, through the stages minus CO - dihapto - minus one more CO - tetrahapto - minus one more CO - hexagapto, so that less than 16 electrons are not obtained.

So, if we have a complex with 16 electrons or less, then the ligand substitution most likely proceeds as an addition-detachment (for lovers of thoughtful words: associative-dissociative or simply associative): the new ligand first comes, then the old one leaves. Two obvious questions arise: why does the old ligand leave, because 18 electrons is very good, and why not do the opposite in this case, as in 18-electron complexes. The first question is easy to answer: each metal has its own habits, and some metals, especially late ones with almost completely filled d-shells, prefer the 16-electron count and the corresponding structural types, and therefore discard the extra ligand, returning to their favorite configuration. Sometimes the spatial factor also interferes with the matter, the already existing ligands are large and the additional one feels like a bus passenger at rush hour. It’s easier to get off and walk around on foot than to suffer like that. However, you can push another passenger out, let him take a walk, and we'll go. The second question is also simple - in this case, the dissociative mechanism would first have to give a 14-electron complex, and this is rarely beneficial.
Here is an example. For a change, we will replace the X-ligand with an L-ligand, and we will not get confused in the oxidation states and charges. Once again: when substituting, the oxidation state does not change, and if the X-ligand is gone, then the loss must be compensated by the charge on the metal. If we forget about this, then the oxidation state would decrease by 1, which is not true.

And one more oddity. A metal-pyridine bond was formed due to the lone pair on nitrogen. In organic chemistry, in this case, we would necessarily show a plus on the nitrogen of pyridine (for example, during protonation or the formation of a quaternary salt), but we never do this in coordination chemistry with either pyridine or any other L-ligands. This is terribly annoying for everyone who is used to the strict and unambiguous system of drawing structures in organic chemistry, but you have to get used to it, it's not that difficult.
And there is no exact analogue of SN2 in the chemistry of coordination compounds, there is a distant one, but it is relatively rare and we do not really need it.
Stable and labile ligands
It would be possible not to talk about the mechanisms of ligand substitution at all, if not for one extremely important circumstance, which we will use a lot: the substitution of ligands, whether associative or dissociative, necessarily implies the dissociation of the old ligand. And it is very important for us to know which ligands leave easily and which leave poorly, preferring to remain in the coordination sphere of the metal.
As we will soon see, in any reaction, some of the ligands remain in the coordination sphere and do not change. Such ligands are usually called spectator ligands (if you do not want such simple, “unscientific” words, use the English word spectator in the local transcription spectator, spectator ligand, but, I beg you, not a spectator - this is unbearable!). And part directly participates in the reaction, turning into reaction products. Such ligands are called actors (not actors!), that is, acting. It is quite clear that the ligand-actors must be easily introduced and removed into the coordination sphere of the metal, otherwise the reaction will simply get stuck. But spectator ligands are better left in the coordination sphere for many reasons, but at least for such a banal one as the need to avoid unnecessary fuss around the metal. It is better that only ligands, actors and in the required quantities, can participate in the desired process. If there are more available coordination sites than necessary, extra ligands-actors can sit on them, and even those that will participate in side reactions, reducing the yield of the target product and selectivity. In addition, spectator ligands almost always perform many important functions, for example, ensure the solubility of complexes, stabilize the correct valence state of the metal, especially if it is not quite usual, help individual steps, provide stereoselectivity, etc. We do not decipher it yet, because we will discuss all this in detail when we get to specific reactions.
It turns out that some of the ligands in the coordination sphere must be strongly bound and not prone to dissociation and substitution by other ligands. Such ligands are called coordinatively stable . Or simply stable, if it is clear from the context that we are talking about the bond strength of the ligands, and not about their own thermodynamic stability, which just does not bother us at all.
And ligands that easily and willingly enter and leave, and are always ready to give way to others, are called coordinatively labile , or simply labile, and here, fortunately, there are no ambiguities.
Cyclobutadiene as a ligand
Here is probably the most striking example of the fact that in the coordination sphere a very unstable molecule can become an excellent ligand, and, by definition, coordinationally stable, if only because if it ventures out of the warm and cozy sphere, nothing good awaits it (at the cost of output is just the energy of anti-aromatic destabilization).
Cyclobutadiene and its derivatives are the best known examples of antiaromaticity. These molecules exist only at low temperatures, and in a highly distorted form - in order to get as far as possible from antiaromaticity, the cycle is distorted into an elongated rectangle, removing delocalization and weakening the conjugation of double bonds as much as possible (otherwise, this is called the Jahn-Teller effect of the 2nd kind: degenerate system, and cyclobutadiene square is a degenerate diradical, remember Frost's circle - distorted and reduces symmetry to remove degeneracy).
But in complexes, cyclobutadiene and substituted cyclobutadienes are excellent tetrahapto ligands, and the geometry of such ligands is precisely a square, with identical bond lengths. How and why this happens is a separate story, and far from being as obvious as it is often presented.
Coordination labile ligands
You need to understand that there is no reinforced concrete fence with barbed wire and guard towers between the areas of labile and stable ligands. First, it depends on the metal, and in this context, GMKO works well. For example, late transition metals prefer soft ligands, while early transition metals prefer hard ligands. For example, iodide clings very tightly to the d 8 atoms of palladium or platinum, but rarely even enters the coordination sphere of titanium or zirconium in the d 0 configuration. But in many metal complexes with not so pronounced features, iodide manifests itself as a completely labile ligand, easily giving way to others.
Other things being equal:
- L-ligands are generally more labile than X-ligands;
- the lability of X-ligands is determined by the hardness/softness and nature of the metal;
- “implicit” ligands are very labile: solvents and bridges in dimers and clusters, so much so that their presence in the coordination sphere is often neglected altogether and structures are drawn without them with a formally unsaturated coordination sphere;
- digapto ligands, such as alkenes and alkynes, behave like typical L ligands: they are usually quite labile;
- ligands with a higher haptiness are rarely labile, but if a poly-hapto ligand can change the mode of bonding to a mono-hapto, it becomes more labile, for example, η 3 -allyls behave this way;
- chelate ligands forming 5- and 6-membered chelate rings are stable, while chelates with fewer or more ring atoms are labile at least at one center (the chelate ring opens and the ligand remains hanging as a simple one). This is how, for example, acetate behaves;
Coordinationally stable ligands
Let's do it again, but on the other side
In the coordination sphere of metals, as a rule, the following are preserved (are coordinately stable):
- 5 and 6-membered chelators;
- polyhapto-ligands: in order to knock cyclopentadienyls or benzene (arenes) out of the coordination sphere, all sorts of special tricks have to be used - they just don’t come out just like that, often withstanding even prolonged heating;
- ligands associated with the metal with a high proportion of the π-donor effect (back-donation);
- soft ligands in late transition metals;
- the “last” ligand in the coordination sphere.
The last condition looks strange, but imagine a complex that has many different ligands, among which there are no unconditionally stable ones (no chelators and polygapto-ligands). Then, in the reactions, the ligands will change, relatively speaking, in the order of relative lability. The least labile and will remain the last. Such a focus occurs, for example, when we use phosphine complexes of palladium. Phosphines are relatively stable ligands, but when there are many of them, and the metal is rich in electrons (d 8 , d 10), they give way, one by one, to ligand-actors. But the last phosphine ligand usually remains in the coordination sphere, and this is very good from the point of view of the reactions in which these complexes participate. We will return to this important issue later. Here is a rather typical example, when only one “last” phosphine remains from the initial coordination sphere of the palladium phosphine complex in the Heck reaction. This example brings us very close to the most important concept in transition metal complex reactions, the concept of ligand control. We'll discuss later.

Remetalization
When substituting one ligand for another, it is important not to overdo it with the reactivity of the incoming ligand. When we are dealing with reactions of organic molecules, it is important for us to deliver exactly one molecule of each of the reagents to the coordination sphere. If two molecules enter instead of one, there is a high probability of side reactions involving two identical ligands. A loss of reactivity is also possible due to the saturation of the coordination sphere and the impossibility of introducing into it other ligands necessary for the expected process. This problem arises especially often when strong anionic nucleophiles, for example, carbanions, are introduced into the coordination sphere. To avoid this, less reactive derivatives are used, in which, instead of an alkali metal cation, which causes a high bond ionicity, less electropositive metals and metalloids (zinc, tin, boron, silicon, etc.) are used that form covalent bonds with the nucleophilic part . Reactions of such derivatives with transition metal derivatives give ligand substitution products, in principle, exactly as if the nucleophile were in the anionic form, but due to reduced nucleophilicity with fewer complications and no side reactions.
Such ligand substitution reactions are usually called transmetallation to emphasize the obvious fact that the nucleophile seems to change metals - more electropositive to less electropositive. Thus, this name contains an element of unpleasant schizophrenia - we seem to have already agreed that we will look at all reactions from the point of view of the transition metal, but suddenly we broke down again and look at this reaction and only this reaction from the point of view of a nucleophile. We'll have to be patient, that's how the terminology has developed and that's how it is accepted. In fact, this word goes back to the early chemistry of organometallic compounds and to the fact that the action of lithium or organomagnesium compounds on the halides of various metals and metalloids is one of the main methods for the synthesis of any organometallic, primarily intransitive, and the reaction that we are now considering in chemistry of coordination compounds of transition metals is simply a generalization of the old method of organometallic chemistry, from which it all grew.
![]()
How does remetallization take place?
Remetaling is both similar to regular substitution and not. It seems that if we consider an intransitive organometallic reagent as just a carbanion with a counterion, then the carbon-intransition metal bond is ionic. But this idea seems to be true only for the most electropositive metals - for magnesium. But already for zinc and tin this idea is very far from the truth.
Therefore, two σ-bonds and four atoms at their ends enter into the reaction. As a result, two new σ-bonds are formed and four atoms are bound to each other in a different order. Most likely, all this occurs simultaneously in a four-membered transition state, and the reaction itself has a concerted character, like very many other reactions of transition metals. The abundance of electrons and orbitals for literally all tastes and all kinds of symmetries makes transition metals capable of simultaneously maintaining bonds in transition states with several atoms.
In the case of remetalization, we obtain a special case of a very general process, which is simply called σ-bond metathesis. Do not confuse only with true metathesis of olefins and acetylenes, which are full-fledged catalytic reactions with their own mechanisms. In this case, we are talking about the mechanism of remetalization or another process in which something similar occurs.

Ligands - ions or molecules that are directly associated with the complexing agent and are donors of electron pairs. These electron-rich systems, which have free and mobile electron pairs, can be electron donors, for example: Compounds of p-elements exhibit complexing properties and act as ligands in a complex compound. Ligands can be atoms and molecules
(protein, amino acids, nucleic acids, carbohydrates). The efficiency and strength of the donor-acceptor interaction between a ligand and a complexing agent is determined by their polarizability, i.e., the ability of a particle to transform its electron shells under external influence.
Instability constant:
Knest= 2 /
K mouth \u003d 1 / Knest
Ligand substitution reactions
One of the most important steps in metal complex catalysis, the interaction of the Y substrate with the complex, proceeds via three mechanisms:
a) Replacement of the ligand with a solvent. Usually such a stage is depicted as the dissociation of the complex
The essence of the process in most cases is the replacement of the ligand L by the solvent S, which is then easily replaced by the substrate molecule Y
b) Attachment of a new ligand along a free coordinate with the formation of an associate, followed by dissociation of the substituted ligand
c) Synchronous substitution (type S N 2) without the formation of an intermediate
Ideas about the structure of metalloenzymes and other biocomplex compounds (hemoglobin, cytochromes, cobalamins). Physical and chemical principles of oxygen transport by hemoglobin.
Structural features of metalloenzymes.
Biocomplex compounds vary considerably in stability. The role of the metal in such complexes is highly specific: replacing it even with an element with similar properties leads to a significant or complete loss of physiological activity.
1. B12: contains 4 pyrrole rings, cobalt ion and CN- groups. Promotes the transfer of the H atom to the C atom in exchange for any group, participates in the formation of deoxyribose from ribose.
2. hemoglobin: has a quaternary structure. Four polypeptide chains connected together form an almost regular ball shape, where each chain contacts two chains.
Hemoglobin is a respiratory pigment that gives blood its red color. Hemoglobin is composed of protein and iron porphyrin and carries oxygen from the respiratory organs to body tissues and carbon dioxide from them to the respiratory organs.
Cytochromes- complex proteins (hemoproteins) that carry out stepwise transfer of electrons and / or hydrogen from oxidizable organic substances to molecular oxygen in living cells. This produces an energy-rich ATP compound.
Cobalamins- natural biologically active organocobalt compounds. The structural basis of cobalt is a corrin ring, consisting of 4 pyrrole nuclei, in which the nitrogen atoms are bonded to the central cobalt atom.
Physico-chemical principles of oxygen transport by hemoglobin- Atom (Fe (II)) (one of the components of hemoglobin) is able to form 6 coordination bonds. Of these, four are used to fix the Fe(II) atom itself in the heme, the fifth bond is used to bind the heme to the protein subunit, and the sixth bond is used to bind the O 2 or CO 2 molecule.
Metal-ligand homeostasis and causes of its violation. Mechanism of toxic action of heavy metals and arsenic based on the theory of hard and soft acids and bases (HMBA). Thermodynamic principles of chelation therapy. Mechanism of cytotoxic action of platinum compounds.
In the body, the formation and destruction of biocomplexes from metal cations and bioligands (porphins, amino acids, proteins, polynucleotides), which include donor atoms of oxygen, nitrogen, and sulfur, continuously occur. The exchange with the environment maintains the concentrations of these substances at a constant level, providing metal ligand homeostasis. Violation of the existing balance leads to a number of pathological phenomena - metal surplus and metal deficiency states. An incomplete list of diseases associated with changes in the metal-ligand balance for only one ion, the copper cation, can be cited as an example. Deficiency of this element in the body causes Menkes syndrome, Morfan syndrome, Wilson-Konovalov disease, cirrhosis of the liver, emphysema, aorto- and arteriopathy, anemia. Excessive intake of the cation can lead to a series of diseases of various organs: rheumatism, bronchial asthma, inflammation of the kidneys and liver, myocardial infarction, etc., called hypercupremia. Professional hypercupreosis is also known - copper fever.
The circulation of heavy metals occurs partially in the form of ions or complexes with amino acids, fatty acids. However, the leading role in the transport of heavy metals belongs to proteins that form a strong bond with them.
They are fixed on cell membranes, block the thiol groups of membrane proteins- 50% of them are protein-enzymes that disrupt the stability of the protein-lipid complexes of the cell membrane and its permeability, causing the release of potassium from the cell and the penetration of sodium and water into it.
A similar effect of these poisons, which are actively fixed on red blood cells, leads to disruption of the integrity of erythrocyte membranes, inhibition of aerobic glycolysis and metabolism processes in them in general, and accumulation of hemolytically active hydrogen peroxide due to inhibition of peroxidase in particular, which leads to the development of one of the characteristic symptoms of poisoning by compounds this group - to hemolysis.
The distribution and deposition of heavy metals and arsenic occur in almost all organs. Of particular interest is the ability of these substances to accumulate in the kidneys, which is explained by the rich content of thiol groups in the renal tissue, the presence of a protein in it - metallobionin, which contains a large number of thiol groups, which contributes to the long-term deposition of poisons. The liver tissue, also rich in thiol groups and containing metallobionin, is also distinguished by a high degree of accumulation of toxic compounds of this group. The term of deposit, for example, of mercury can reach 2 months or more.
The excretion of heavy metals and arsenic occurs in different proportions through the kidneys, liver (with bile), mucous membrane of the stomach and intestines (with feces), sweat and salivary glands, lungs, which is usually accompanied by damage to the excretory apparatus of these organs and manifests itself in the corresponding clinical symptoms.
The lethal dose for soluble mercury compounds is 0.5 g, for calomel 1–2 g, for copper sulfate 10 g, for lead acetate 50 g, for white lead 20 g, for arsenic 0.1–0.2 g.
The concentration of mercury in the blood is more than 10 µg/l (1γ%), in the urine more than 100 µg/l (10γ%), the concentration of copper in the blood is more than 1600 µg/l (160γ%), arsenic is more than 250 µg/l (25γ%) %) in the urine.
Chelation therapy is the removal of toxic particles
from the body, based on their chelation
s-element complexonates.
Drugs used to remove
incorporated in the body of toxic
particles are called detoxifiers.
Chapter 17
17.1. Basic definitions
In this chapter, you will be introduced to a special group of complex substances called comprehensive(or coordinating) connections.
Currently, a strict definition of the concept " complex particle" no. The following definition is usually used.
For example, a hydrated copper ion 2 is a complex particle, since it actually exists in solutions and some crystalline hydrates, it is formed from Cu 2 ions and H 2 O molecules, water molecules are real molecules, and Cu 2 ions exist in crystals of many copper compounds. On the contrary, the SO 4 2 ion is not a complex particle, since although O 2 ions occur in crystals, the S 6 ion does not exist in chemical systems.
Examples of other complex particles: 2 , 3 , , 2 .
At the same time, NH 4 and H 3 O ions are classified as complex particles, although H ions do not exist in chemical systems.
Sometimes complex particles are called complex chemical particles, all or part of the bonds in which are formed according to the donor-acceptor mechanism. This is true in most complex particles, but, for example, in potassium alum SO 4 in complex particle 3, the bond between Al and O atoms is indeed formed according to the donor-acceptor mechanism, while in the complex particle there is only electrostatic (ion-dipole) interaction. This is confirmed by the existence in iron ammonium alum of a complex particle similar in structure, in which only ion-dipole interaction is possible between water molecules and the NH 4 ion.
By charge, complex particles can be cations, anions, and also neutral molecules. Complex compounds containing such particles can belong to different classes of chemicals (acids, bases, salts). Examples: (H 3 O) - acid, OH - base, NH 4 Cl and K 3 - salts.
Typically, the complexing agent is an atom of an element that forms a metal, but it can also be an atom of oxygen, nitrogen, sulfur, iodine, and other elements that form non-metals. The oxidation state of the complexing agent may be positive, negative, or zero; when a complex compound is formed from simpler substances, it does not change.
Ligands can be particles that, before the formation of a complex compound, were molecules (H 2 O, CO, NH 3, etc.), anions (OH, Cl, PO 4 3, etc.), as well as a hydrogen cation. Distinguish unidentate or monodentate ligands (linked to the central atom through one of its atoms, that is, by one -bond), bidentate(connected to the central atom through two of their atoms, that is, by two -bonds), tridentate etc.
If the ligands are unidentate, then the coordination number is equal to the number of such ligands.
The cn depends on the electronic structure of the central atom, its degree of oxidation, the size of the central atom and ligands, the conditions for the formation of the complex compound, temperature, and other factors. CN can take values from 2 to 12. Most often it is equal to six, somewhat less often - four.
There are also complex particles with several central atoms.
Two types of structural formulas of complex particles are used: indicating the formal charge of the central atom and ligands, or indicating the formal charge of the entire complex particle. Examples:

To characterize the shape of a complex particle, the idea of a coordination polyhedron (polyhedron) is used.

Coordination polyhedra also include a square (KN = 4), a triangle (KN = 3), and a dumbbell (KN = 2), although these figures are not polyhedra. Examples of coordination polyhedra and correspondingly shaped complex particles for the most common CN values are shown in Figs. one.
17.2. Classification of complex compounds
How chemicals complex compounds are divided into ionic (they are sometimes called ionogenic) and molecular ( non-ionic) connections. Ionic complex compounds contain charged complex particles - ions - and are acids, bases or salts (see § 1). Molecular complex compounds consist of uncharged complex particles (molecules), for example: or - it is difficult to assign them to any main class of chemicals.
The complex particles that make up complex compounds are quite diverse. Therefore, several classification features are used for their classification: the number of central atoms, the type of ligand, the coordination number, and others.
According to the number of central atoms complex particles are divided into single-core and multi-core. The central atoms of multinuclear complex particles can be linked to each other either directly or through ligands. In both cases, the central atoms with ligands form a single inner sphere of the complex compound:
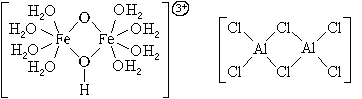

According to the type of ligands, complex particles are divided into
1) Aquacomplexes, that is, complex particles in which water molecules are present as ligands. Cationic aquacomplexes m are more or less stable, anionic aquacomplexes are unstable. All crystalline hydrates are compounds containing aqua complexes, for example:
Mg(ClO 4) 2. 6H 2 O is actually (ClO 4) 2 ;
BeSO4. 4H 2 O is actually SO 4 ;
Zn(BrO 3) 2 . 6H 2 O is actually (BrO 3) 2 ;
CuSO4. 5H 2 O is actually SO 4 . H2O.
2) Hydroxocomplexes, that is, complex particles in which hydroxyl groups are present as ligands, which were hydroxide ions before entering the complex particle, for example: 2 , 3 , .
Hydroxo complexes are formed from aqua complexes that exhibit the properties of cationic acids:
2 + 4OH = 2 + 4H 2 O
3) Ammonia, that is, complex particles in which NH 3 groups are present as ligands (before the formation of a complex particle - ammonia molecules), for example: 2 , , 3 .
Ammonia can also be obtained from aqua complexes, for example:
2 + 4NH 3 \u003d 2 + 4 H 2 O
The color of the solution in this case changes from blue to ultramarine.
4) acidocomplexes, that is, complex particles in which acidic residues of both oxygen-free and oxygen-containing acids are present as ligands (before the formation of a complex particle - anions, for example: Cl, Br, I, CN, S 2, NO 2, S 2 O 3 2 , CO 3 2 , C 2 O 4 2 etc.).
Examples of the formation of acid complexes:
Hg 2 + 4I = 2
AgBr + 2S 2 O 3 2 = 3 + Br
The latter reaction is used in photography to remove unreacted silver bromide from photographic materials.
(When developing photographic film and photographic paper, the unexposed part of the silver bromide contained in the photographic emulsion is not restored by the developer. To remove it, this reaction is used (the process is called "fixing", since the unremoved silver bromide gradually decomposes in the light, destroying the image)
5) Complexes in which hydrogen atoms are ligands are divided into two completely different groups: hydride complexes and complexes included in the composition onium connections.
In the formation of hydride complexes - , , - the central atom is an electron acceptor, and the hydride ion is a donor. The oxidation state of hydrogen atoms in these complexes is –1.
In onium complexes, the central atom is an electron donor, and the acceptor is a hydrogen atom in the +1 oxidation state. Examples: H 3 O or - oxonium ion, NH 4 or - ammonium ion. In addition, there are substituted derivatives of such ions: - tetramethylammonium ion, - tetraphenylarsonium ion, - diethyloxonium ion, etc.
6) Carbonyl complexes - complexes in which CO groups are present as ligands (before complex formation - carbon monoxide molecules), for example:,, etc.
7) Anion halide complexes are complexes of type .
Other classes of complex particles are also distinguished according to the type of ligands. In addition, there are complex particles with ligands of various types; the simplest example is aqua hydroxocomplex.
17.3. Fundamentals of the nomenclature of complex compounds
The formula of a complex compound is compiled in the same way as the formula of any ionic substance: the formula of the cation is written in the first place, and the anion in the second.
The formula of a complex particle is written in square brackets in the following sequence: the symbol of the complexing element is placed first, then the formulas of the ligands that were cations before the formation of the complex, then the formulas of the ligands that were neutral molecules before the formation of the complex, and after them the formulas of the ligands, former before the formation of the complex by anions.
The name of a complex compound is built in the same way as the name of any salt or base (complex acids are called hydrogen or oxonium salts). The name of the compound includes the name of the cation and the name of the anion.
The name of the complex particle includes the name of the complexing agent and the names of the ligands (the name is written in accordance with the formula, but from right to left. For complexing agents in cations, Russian element names are used, and in anions, Latin ones.
Names of the most common ligands:
| H 2 O - aqua | Cl - chloro | SO 4 2 - sulfate | OH - hydroxo |
| CO - carbonyl | Br - bromo | CO 3 2 - carbonate | H - hydrido |
| NH 3 - ammine | NO 2 - nitro | CN - cyano | NO - nitroso |
| NO - nitrosyl | O 2 - oxo | NCS - thiocyanato | H + I - hydro |
Examples of names of complex cations:
Examples of names of complex anions:
2 - tetrahydroxozincate ion
3 – di(thiosulfato)argentate(I)-ion
3 – hexacyanochromate(III)-ion
– tetrahydroxodiquaaluminate ion
– tetranitrodiamminecobaltate(III)-ion
3 – pentacyanoaquaferrate(II)-ion
Examples of the names of neutral complex particles:
More detailed nomenclature rules are given in reference books and special manuals.
17.4. Chemical bond in complex compounds and their structure
In crystalline complex compounds with charged complexes, the bond between the complex and the outer sphere ions is ionic, while the bonds between the remaining particles of the outer sphere are intermolecular (including hydrogen bonds). In molecular complex compounds, the bond between the complexes is intermolecular.
In most complex particles, the bonds between the central atom and the ligands are covalent. All or part of them are formed according to the donor-acceptor mechanism (as a result, with a change in formal charges). In the least stable complexes (for example, in the aqua complexes of alkaline and alkaline earth elements, as well as ammonium), ligands are held by electrostatic attraction. The bond in complex particles is often referred to as a donor-acceptor or coordination bond.
Let us consider its formation using the iron(II) aquacation as an example. This ion is formed by the reaction:
FeCl 2cr + 6H 2 O = 2 + 2Cl
The electronic formula of the iron atom is 1 s 2 2s 2 2p 6 3s 2 3p 6 4s 2 3d 6. Let's make a scheme of valence sublevels of this atom:
When a doubly charged ion is formed, the iron atom loses two 4 s-electron:
The iron ion accepts six electron pairs of oxygen atoms of six water molecules into free valence orbitals:

A complex cation is formed, the chemical structure of which can be expressed by one of the following formulas:

The spatial structure of this particle is expressed by one of the spatial formulas:

The shape of the coordination polyhedron is an octahedron. All Fe-O bonds are the same. Supposed sp 3 d 2 - hybridization of iron atom AO. The magnetic properties of the complex indicate the presence of unpaired electrons.
If FeCl 2 is dissolved in a solution containing cyanide ions, then the reaction proceeds
FeCl 2cr + 6CN = 4 + 2Cl.
The same complex is also obtained by adding a solution of potassium cyanide KCN to a FeCl 2 solution:
2 + 6CN \u003d 4 + 6H 2 O.
This suggests that the cyanide complex is stronger than the aquacomplex. In addition, the magnetic properties of the cyanide complex indicate the absence of unpaired electrons from the iron atom. All this is due to a slightly different electronic structure of this complex:

The "stronger" CN ligands form stronger bonds with the iron atom, the energy gain is enough to "break" the Hund's rule and release 3 d-orbitals for lone pairs of ligands. The spatial structure of the cyanide complex is the same as that of the aquacomplex, but the type of hybridization is different - d 2 sp 3 .
The "strength" of the ligand depends primarily on the electron density of the cloud of the lone pair of electrons, that is, it increases with a decrease in the size of the atom, with a decrease in the principal quantum number, depends on the type of EO hybridization and on some other factors. The most important ligands can be lined up in order of increasing their "strength" (a kind of "activity series" of ligands), this series is called spectrochemical series of ligands:
| I; Br; : SCN, Cl, F, OH, H 2 O; : NCS, NH3; SO 3 S : 2 ; : CN, CO |
For complexes 3 and 3, the formation schemes look as follows:
|
|


For complexes with CN = 4, two structures are possible: a tetrahedron (in the case sp 3-hybridization), for example, 2 , and a flat square (in the case of dsp 2 hybridization), for example, 2 .
17.5. Chemical properties of complex compounds
For complex compounds, first of all, the same properties are characteristic as for ordinary compounds of the same classes (salts, acids, bases).
If the compound is an acid, then it is a strong acid; if it is a base, then the base is strong. These properties of complex compounds are determined only by the presence of H 3 O or OH ions. In addition, complex acids, bases and salts enter into the usual exchange reactions, for example:
SO 4 + BaCl 2 \u003d BaSO 4 + Cl 2
FeCl 3 + K 4 = Fe 4 3 + 3KCl
The last of these reactions is used as a qualitative reaction for Fe 3 ions. The resulting ultramarine insoluble substance is called "prussian blue" [the systematic name is iron(III)-potassium hexacyanoferrate(II)].
In addition, the complex particle itself can enter into the reaction, and the more actively, the less stable it is. Usually these are ligand substitution reactions occurring in solution, for example:
2 + 4NH 3 \u003d 2 + 4H 2 O,
as well as acid-base reactions such as
2 + 2H 3 O = + 2H 2 O
2 + 2OH = + 2H 2 O
Formed in these reactions, after isolation and drying, it turns into zinc hydroxide:
Zn(OH) 2 + 2H 2 O
The last reaction is the simplest example of the decomposition of a complex compound. In this case, it runs at room temperature. Other complex compounds decompose when heated, for example:
SO4. H 2 O \u003d CuSO 4 + 4NH 3 + H 2 O (above 300 o C)
4K 3 \u003d 12KNO 2 + 4CoO + 4NO + 8NO 2 (above 200 o C)
K 2 \u003d K 2 ZnO 2 + 2H 2 O (above 100 o C)
To assess the possibility of a ligand substitution reaction, a spectrochemical series can be used, guided by the fact that stronger ligands displace weaker ones from the inner sphere.
17.6. Isomerism of complex compounds
Isomerism of complex compounds is related
1) with possible different arrangement of ligands and outer-sphere particles,
2) with a different structure of the most complex particle.
The first group includes hydrated(in general solvate) and ionization isomerism, to the second - spatial and optical.
Hydrate isomerism is associated with the possibility of different distribution of water molecules in the outer and inner spheres of the complex compound, for example: (red-brown color) and Br 2 (blue color).
Ionization isomerism is associated with the possibility of different distribution of ions in the outer and inner spheres, for example: SO 4 (purple) and Br (red). The first of these compounds forms a precipitate, reacting with a solution of barium chloride, and the second - with a solution of silver nitrate.
Spatial (geometric) isomerism, otherwise called cis-trans isomerism, is characteristic of square and octahedral complexes (it is impossible for tetrahedral ones). Example: cis-trans square complex isomerism

Optical (mirror) isomerism essentially does not differ from optical isomerism in organic chemistry and is characteristic of tetrahedral and octahedral complexes (impossible for square ones).
complex compounds. Their structure is based on the coordination theory of A. Werner. Complex ion, its charge. Cationic, anionic, neutral complexes. Nomenclature, examples.



Ligand substitution reactions. Complex ion instability constant, stability constant.

 To instability is the ratio of the products of the concentration of decayed ions to the undecayed amount.
To instability is the ratio of the products of the concentration of decayed ions to the undecayed amount.
K set \u003d 1 / K nest (reciprocal)
 Secondary dissociation - the disintegration of the inner sphere of the complex into its constituent components.
Secondary dissociation - the disintegration of the inner sphere of the complex into its constituent components.
43. Competition for a ligand or for a complexing agent: isolated and combined equilibria of ligand substitution. General constant of combined equilibrium of ligand substitution.
As a result of competition, the proton destroys a sufficiently strong complex, forming a weakly dissociating substance - water.
Cl + NiS0 4 +4NH 3 ^ S0 4 + AgCl I
This is already an example of ligand competition for a complexing agent, with the formation of a more stable complex (K H + \u003d 9.3-1 (G 8; K H [M (W 3) 6 ] 2+ \u003d 1.9-10 -9) and a sparingly soluble compound AgCl - K s \u003d 1.8 10 "10
Ideas about the structure of metalloenzymes and other biocomplex compounds (hemoglobin, cytochromes, cobalamins). Physico-chemical principles of oxygen transport by hemoglobin



Cobalamins. Vitamin B 12 called a group of cobalt-containing biologically active substances called cobalamins. They are actually cyanocobalamin, hydroxycobalamin and two coenzymatic forms of vitamin B 12: methylcobalamin and 5-deoxyadenosylcobalamin.
Sometimes, in a narrower sense, vitamin B 12 is called cyanocobalamin, since it is in this form that the main amount of vitamin B 12 enters the human body, without losing sight of the fact that it is not synonymous with B 12, and several other compounds also have B 12 - vitamin activity. Vitamin B 12 is also called Castle's extrinsic factor.
B 12 has the most complex chemical structure compared to other vitamins, the basis of which is the corrin ring. Corrin is in many ways similar to porphyrin (a complex chemical structure that is part of heme, chlorophyll and cytochromes), but differs from porphyrin in that two pyrrole rings in the composition of corrin are directly connected to each other, and not by a methylene bridge. The cobalt ion is located in the center of the corrin structure. Cobalt forms four coordination bonds with nitrogen atoms. Another coordination bond connects cobalt with dimethylbenzimidazole nucleotide. The last, sixth coordination bond of cobalt remains free: it is through this bond that the cyano group, hydroxyl group, methyl or 5 "-deoxyadenosyl residue is added to form four variants of vitamin B 12, respectively. The carbon-covalent covalent bond in the structure of cyanocobalamin is the only one known in living nature is an example of a covalent bond transition metal-carbon.
