Fundamentals of adsorption thermodynamics. Thermodynamics of adsorption processes
In the case of the interaction of two atoms:
U is the interaction energy;
U = U + U PULL.
 - Lennard-Jones equation
, c, b, m = const
- Lennard-Jones equation
, c, b, m = const
In cases of interaction of atoms with a solid surface, it is necessary to sum up all interactions.
x is the distance to the surface
r - the radius of action of the forces of attraction
dV - volume
n is the number of surface molecules
U ADS. is the adsorption interaction energy



In the case of adsorption, attraction is enhanced. And in the case of interaction of the nonpolar-nonpolar type, adsorption is predominantly localized in the depressions.
electrostatic interaction.
Polar adsorbent - non-polar adsorbate
Non-polar adsorbent - polar adsorbate
A polar adsorbent is a polar adsorbate.
M  the adsorbate molecule is represented as a dipole, and the adsorbent as a conductor, in which the adsorbate molecule induces a dipole mirror-symmetrically with respect to the given one.
the adsorbate molecule is represented as a dipole, and the adsorbent as a conductor, in which the adsorbate molecule induces a dipole mirror-symmetrically with respect to the given one.
X - distance to the middle
When interacting, the potential arises:
 ,
,
 is the dipole moment.
is the dipole moment.
The potential tends to take on the maximum value, i.e. dipoles tend to orient themselves perpendicular to the surface.
Since an increase in temperature promotes the growth of Brownian motion, it leads to a deceleration of the adsorption process.
In the case of electrostatic interaction, the adsorbate is predominantly localized on protrusions.
Fundamental adsorption equation.
In the case of adsorption, the component is redistributed, which means that the chemical potential changes. The process of adsorption can be considered as the transition of surface energy into chemical energy.
Layer volume = 0, then the generalized equation I and II of the law of thermodynamics:
T = const; (1) = (2) => 

For a two-component system:
,
 ,
,
 =>
=>
=>
 - Gibbs adsorption equation
.
- Gibbs adsorption equation
.
For the case of adsorption of TV. body - gas:, 
 ,
,

 - isotherm
- isotherm
 - isobar
- isobar
 - isopykne
- isopykne
 - isostere
- isostere
Isotherm, isopycne, isostere are related to each other.
Because adsorption function 



Henry isotherm Langmuir isotherm



Thermodynamics. Adsorption.
For condensed media:
 ,
,
 ,
,
 - integral change in the Gibbs energy
.
- integral change in the Gibbs energy
.

P-pressure over a curved surface, P S-pressure over a flat surface
 - adsorption potential
- adsorption potential
Differential change in entrapy 
 , Г = const
, Г = const
- differential entropy change
- differential enthalpy of adsorption
 - isosteric heat of adsorption
- isosteric heat of adsorption
 - heat of condensation
- heat of condensation
 - net heat of adsorption
- net heat of adsorption

 ,
,




Qa is the integral heat of adsorption, 
Qra is the integral net heat of adsorption,

Henry's equation
The study of adsorption is hampered by the inhomogeneity of the surface, so the simplest regularities are obtained for homogeneous surfaces.
Let us consider the interaction of gases with a solid surface, when a gas passes from an equilibrium state in the volume to an equilibrium state on the surface. This case is analogous to the equilibrium of gases in a gravitational field.
 ,
,
 ,
=>
,
=> -Henry's equation
-Henry's equation
 - distribution coefficient
- distribution coefficient
In the process of adsorption, a change in chemical potentials occurs.
For bulk phase: 
For surface gas: 
In a state of equilibrium  , i.e.
, i.e.

In the Henry equation, the constant does not depend on the concentration
The Henry equation is valid in the region of low pressures and concentrations. As the concentration increases, 2 types of deviations from Henry's law are possible:
1 - positive deviations, D decreases, A decreases
2 - negative deviations, D - increases, A - increases.
The type of deviation is determined by the predominance of one or another type of adsorbent-adsorbate interaction.
With a strong adhesive interaction, the activity coefficients increase - a positive deviation. In the case of cohesive interactions, negative deviations are observed.
monomolecular adsorption.
Langmuir isotherm.
The simplest regularities were obtained in Henry's theory. Langmuir proposed a theory according to which adsorption is considered as a quasi-chemical reaction. Wherein:
The surface is energetically uniform.
Adsorption is localized, each adsorption center interacts with one adsorbate molecule.
Adsorbate molecules do not interact with each other.
Adsorption is monolayer.

 - surface,
- surface,  - adsorbate,
- adsorbate,  - adsorption complex.
- adsorption complex.
 , then the concentration of adsorption sites:
, then the concentration of adsorption sites:  ,
, - limiting adsorption.
- limiting adsorption.
 , then the reaction constant:
, then the reaction constant: 

 - Langmuir equation.
- Langmuir equation.
Adsorption versus Concentration
1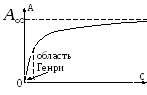 )
)
 ,
,
2) area of high concentrations
 - limiting adsorption, formation of a monomolecular layer
- limiting adsorption, formation of a monomolecular layer
For the Gibbs energy: .
g is the entropy factor.
In the case of the Henry isotherm, the Gibbs energy characterizes the transition of the adsorbate from the standard state in the bulk to the standard state on the surface. In the case of the Langmuir isotherm  characterizes the degree of affinity of the adsorbent and adsorbate.
characterizes the degree of affinity of the adsorbent and adsorbate.
 found from the van't Hoff isobar.
found from the van't Hoff isobar.

 , then
, then  , hence
, hence  .
.

 - degree of surface filling.
- degree of surface filling.




 - the number of vacancies,
- the number of vacancies,  - the number of occupied places.
- the number of occupied places.
 ,
,

Those. in the region of high concentrations, the number of free sites is inversely proportional to the amount of adsorbate.
Adsorption of a mixture of gases on a homogeneous surface.
In this case, the adsorption process is considered as two parallel reactions.
 (1)
(1)

 (2)
(2)


Adsorption of a mixture of gases on an inhomogeneous surface.
In the case of a non-homogeneous surface, one should not limit oneself to medium infills.
As a result of competition, localization of various adsorbates is possible in different types of sites.
In this case, the relation  .
.
 ,
,
 is the saturation vapor pressure of the adsorbate.
is the saturation vapor pressure of the adsorbate.
 ,
,
 is the heat of adsorption.
is the heat of adsorption.
"+" - symbatic dependence, "-" - antibatic dependence, "H" - no correlation.
"+" - adsorption proceeds according to the same mechanism. In the most energetically favorable areas, a gas with a high affinity to the surface is predominantly adsorbed.
"-" - adsorption proceeds through various mechanisms and up to a certain point in time there is no competition for the surface.
Monomolecular adsorption is predominantly realized during the physical adsorption of gases at low values p, as well as at the liquid/gas interface.
Polymolecular adsorption.
BET theory(Brunauer, Emmet, Teller).
In the case when the formation of a monolayer is insufficient to compensate for the surface energy, adsorption is polymolecular and can be considered as a result of forced condensation under the action of surface forces.
Basic provisions:
When an adsorbate molecule hits the occupied site, a multiple set is formed.
As you get closer p to p s the number of free adsorption sites decreases. Initially, the number of places occupied by singles, doubles, etc. increases and then decreases. kits.
At p =p s adsorption turns into condensation.
There are no horizontal interactions.
For the first layer, the Langmuir isotherm is performed.
The surface is considered as a set of adsorption sites. The condition of dynamic equilibrium is valid: the rate of condensation in free places is equal to the rate of evaporation from occupied ones.
a is the condensation coefficient (the fraction of molecules condensed on the surface);
 ,
,
Zm is the maximum number of free places.
 - frequency of vibrations of atoms in the direction perpendicular to the surface.
- frequency of vibrations of atoms in the direction perpendicular to the surface.
For the first layer, the dynamic equilibrium conditions are:



 , then
, then
 - Langmuir equation.
- Langmuir equation.
For the second layer will be true: 
For the i-th layer: 
For simplicity, it is assumed that a and ν are the same for all layers except the first one. For all layers except the first, the heat of adsorption is constant. For the last layer, the heat of adsorption is equal to the heat of condensation. As a result, the equation
 (*)
(*)
C- constant,
In the case of BET theory, the constant FROM characterizes the Gibbs energy of pure adsorption. The equation contains only one constant, and this equation is also very important for determining the specific surface area of the adsorbent.
Since heat is released as a result of adsorption, the determination of specific surfaces is carried out at low temperatures.
 ????????????
????????????


The main flaw of the theory– Neglect of horizontal interactions in favor of vertical ones.
The equation is in the range  from 0.05 to 0.3.
from 0.05 to 0.3.
Where  <
0,05 – существенное влияние оказывает
неоднородность поверхности.
<
0,05 – существенное влияние оказывает
неоднородность поверхности.
 > 0.3 - the interaction of the adsorbate - adsorbate affects.
> 0.3 - the interaction of the adsorbate - adsorbate affects.
Accounting for adsorbate-adsorbate interactions.
Interactions appear during adsorption on a nonpolar surface of branched molecules or molecules. capable of forming associations. In this case, the shape of the adsorption isotherms changes.
BUT  the sorbent is not polar.
the sorbent is not polar.
Graph 1 corresponds to weak interactions adsorbate-adsorbate, strong adsorbate-adsorbent.
Graph 2 corresponds to a strong adsorbate-adsorbate interaction, a strong adsorbate-adsorbent interaction.
Graph 3 corresponds to a strong adsorbate-adsorbate interaction, a weak adsorbate-adsorbent interaction.
 ,
,

In the case of interaction between adsorbate molecules, it is necessary to take into account changes in the activity coefficients. And this equation is written as:
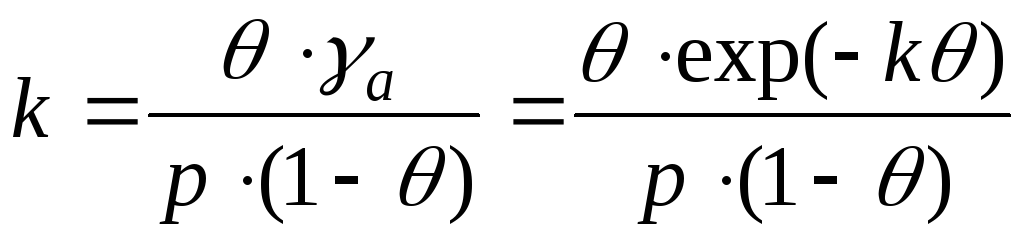 - the equation of Frunkin, Fowler, Guggenheim.
- the equation of Frunkin, Fowler, Guggenheim.
k is the attraction constant.
Polan's potential theory.
This theory does not derive any type of adsorption isotherm, but makes it possible to calculate isotherms at a different temperature.
Adsorption is the result of the attraction of the adsorbate to the surface of the adsorbent due to the action of the adsorption potential, which does not depend on the presence of other molecules and depends on the distance between the surface and the adsorbate molecule.
 ,
,
 - adsorption potential.
- adsorption potential.
Since the surface is inhomogeneous, the distance is replaced by the adsorption volume  .adsorption volume is the volume enclosed between the surface and the point corresponding to the given value
.adsorption volume is the volume enclosed between the surface and the point corresponding to the given value  .
.
Adsorption potential is the work of transferring 1 mol of the adsorbate outside the given adsorption volume to a given point of the adsorption volume (or the work of transferring 1 mol of saturated vapor of the adsorbate, which is in equilibrium with the liquid adsorbate in the absence of the adsorbent, into the vapor phase in equilibrium with the adsorbent).

Characteristic curve
 - adsorption potential,
- adsorption potential, 
For a given adsorbent and various adsorbates, the following is true: 
For different types of adsorbates  ,
,
where  potentials for adsorption isotherms at relative pressures
potentials for adsorption isotherms at relative pressures  for adsorbate 1 and for adsorbate 2. This ratio is a constant value.
for adsorbate 1 and for adsorbate 2. This ratio is a constant value.
 - affinity coefficient
- affinity coefficient
Theory of capillary condensation.
The course of the adsorption process largely depends on the structure of the porous body.
|
microporous | |
|
Transitional porous | |
|
Macroporous |
In the case of microporous sorbents, the fields of adsorption forces overlap. In the case of macroporous sorbents, the pores act as transport channels. The processes of condensation are most significant in transient porous bodies. Capillary condensation starts at certain values p and  when part of the surface energy has already been compensated. A necessary condition is that the surface must be self-supporting. The process is described Thompson-Kelvin equation.
when part of the surface energy has already been compensated. A necessary condition is that the surface must be self-supporting. The process is described Thompson-Kelvin equation.

 - for the case of wetting, the center of curvature is in the gas phase.
- for the case of wetting, the center of curvature is in the gas phase.
In the case of capillary condensation, the adsorption isotherm has a hysteresis form. The lower branch corresponds to the adsorption process, and the upper branch corresponds to the desorption process.
All types of pores can be reduced to three types:
|
conical |
Cylindrical with one closed end |
Cylindrical with two open ends |
|
Process filling is carried out from the bottom of the pore. The adsorption isotherm and the desorption isotherm in this case coincide, since the adsorption process begins with a sphere and the desorption process also begins with the disappearance of some spheres.
↓ |
There is no hysteresis. Forward and reverse stroke are described by the equation:
|
There is no bottom anywhere, the filling of the pore will go along the walls of the cylinder.
cylinder: Isotherm and will have a hysteresis form.
↓ |


 - sphere,
- sphere, ,
,



AT  wetting conditions, condensation occurs at lower pressures, which is energetically favorable. From the desorption branch, pore size distribution curves are obtained.
wetting conditions, condensation occurs at lower pressures, which is energetically favorable. From the desorption branch, pore size distribution curves are obtained.
The maximum of the differential curve is shifted to the left relative to the inflection point of the integral. The total volume of small pores is small, but has large surface areas. As the pore size increases, their volume increases as  , and the area as
, and the area as  , due to this, a shift of the maximum of the differential curve is observed.
, due to this, a shift of the maximum of the differential curve is observed.
Adsorption at the solid-liquid interface.
In the case of adsorption at the solid-gas interface, we neglected one component. In the case of adsorption at the solid-liquid interface, the adsorbate displaces solvent molecules from the surface of the adsorbent.
 ,
,
The right equation is:

 ,
,
N 1, N 2 - mole fractions of the solvent and component, N 1 + N 2 \u003d 1, then
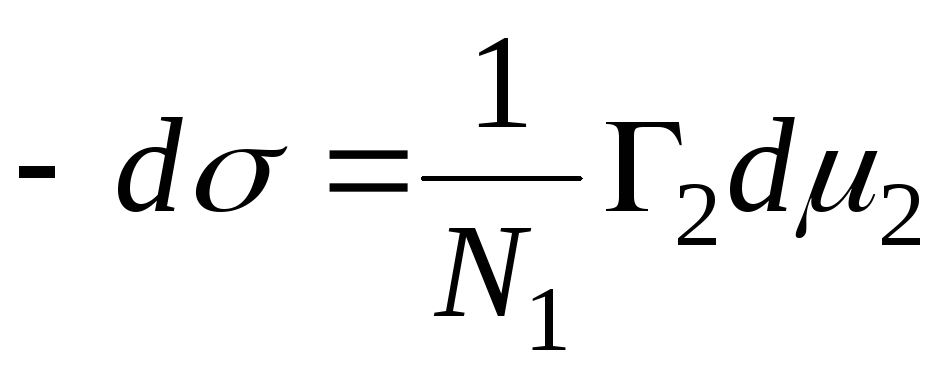 ,
=>
,
=>
 , then - the adsorption equation for the phase boundary solid - liquid.
, then - the adsorption equation for the phase boundary solid - liquid.
Adsorption (G) > 0 at  <
0
<
0
If the values  for the component and solvent are very different, in this case the dependence G from N has an extremum at the value N
~ 0,5.
for the component and solvent are very different, in this case the dependence G from N has an extremum at the value N
~ 0,5.
E  if
if  have similar values, in this case the sign of adsorption may change. Addiction G from N crosses the x-axis
have similar values, in this case the sign of adsorption may change. Addiction G from N crosses the x-axis
Function Intersection G(N) with the abscissa axis is called adsorption azeotrope. This means that the two components cannot be separated on this adsorbent.
Adsorption isotherm equation with exchange constant.
During adsorption at the solid-liquid interface, the components are constantly redistributed between the surface of the adsorbent and the volume of the solution.
 - components (- - refer to the surface)
- components (- - refer to the surface)

 ,
,
 ,
, .
.
 ,
,


Adsorption at the liquid-gas interface
R  Let us consider the change in the concentration profile as the liquid-gas interface is crossed. Let component 2 be volatile.
Let us consider the change in the concentration profile as the liquid-gas interface is crossed. Let component 2 be volatile.
Cs is the concentration in the surface layer.
Based on the definition of excess adsorption

If the component is not volatile, then the adsorption value will be written as follows:
P  ri
ri 

In the equation  the nature of matter is described by the derivative
the nature of matter is described by the derivative  .
.
The surface tension isotherm can be of the form 1 or 2:
1 - surfactants
2 - surfactants
Surface activity g is the ability of substances to reduce surface tension in the system.

 - thickness of the surface layer
- thickness of the surface layer
C s is the concentration of the component in the surface layer
FROM– volume concentration
For a homologous series there is a rule:
 - Traubeau Duclos rule
- Traubeau Duclos rule
For the homologous series, the adsorption isotherm looks like this:
We write D instead of A, since adsorption is excessive in the surface layer.
Surface Tension Isotherm:

 is the surface tension of the pure solvent.
is the surface tension of the pure solvent.
 - fundamental adsorption equation;
- fundamental adsorption equation;
 - Langmuir equation.
- Langmuir equation.
Let's solve them together:

- Shishkovsky's equation.
B is a constant for the homologous series.
A- when moving from one homologue to another, it increases by 3-3.5 times 
![]()
1 - area of low concentrations
![]()
2 - average concentration
3 - monomolecular layer
Surfactants are amphiphilic molecules, i.e. include a polar group and a non-polar hydrocarbon radical.
o is the polar part of the molecule.
| is the non-polar part of the molecule.
In a polar solvent, surfactant molecules are oriented in such a way that the polar part of the molecule faces the solvent, while the nonpolar part is pushed into the gas phase.
In the Shishkovsky equation  , it is constant for the homologous series.
, it is constant for the homologous series.
Surface-active action begins to appear with n>5. At concentrations higher than the concentration of the monomolecular layer, micellization occurs in surfactant solutions.
Micelle- the aggregate of amphiphilic surfactant molecules is called, the hydrocarbon radicals of which form the core, and the polar groups are turned into the aqueous phase.
Micelle mass - micellar mass.
H  the number of molecules is the number of aggregation.
the number of molecules is the number of aggregation.
Spherical micelles
In the case of micellization, an equilibrium is established in the solution
CMC is the critical micelle concentration.
Since we consider the micelle to be a separate phase:
For the homological series, there is an empirical equation:
a is the dissolution energy of the functional group.
b is the adsorption potential increment, the work of adsorption per methylene unit.
is the adsorption potential increment, the work of adsorption per methylene unit.
The presence of a hydrocarbon core in micelles makes it possible for compounds that are insoluble in water to dissolve in aqueous solutions of surfactants, this phenomenon is called solubilization (what dissolves is a solubilizate, surfactant is a solubilizer).
The mud may be completely non-polar, may contain both polar and non-polar parts, and will be oriented like a surfactant molecule.
In any case, during solubilization, an increase in the micellar mass and aggregation number occurs not only due to the inclusion of the solubilizate, but also due to an increase in the number of surfactant molecules necessary to maintain the equilibrium state.
Solubilization is the more effective, the lower the molecular weight of the solubilizate.

 ~ 72 mN/m.
~ 72 mN/m.
 ~ 33 mN/m.
~ 33 mN/m.
The effectiveness of surfactants depends on the magnitude of the CMC.
2D Surface Layer Pressure

→ -forces of surface tension.
- two-dimensional pressure.
The surface layer is a force equal to the difference between the surface tensions of a surfactant solution and a pure solvent, directed towards a clean surface.
An equilibrium is established between the solution and the surface layer



At  there is an area where
there is an area where  linearly dependent on concentration.
linearly dependent on concentration.
G [mol / m 2].
 area occupied by one mole of a substance
area occupied by one mole of a substance
Then the two-dimensional pressure isotherm will have the form 
 is the two-dimensional pressure isotherm.
is the two-dimensional pressure isotherm.
Addiction  from S M:
from S M:

At  - two-dimensional pressure increases sharply. At
- two-dimensional pressure increases sharply. At  two-dimensional is deformed, which causes a sharp growth
two-dimensional is deformed, which causes a sharp growth  .
.
The film on both sides limited by the same phases is called double-sided. In such films, a constant movement of the mother liquor is observed.
Films less than 5 nm thick are called black films.

Adsorption layers should have two characteristics: viscosity and easy mobility, fluidity and elasticity.
The Marangoni effect is self-healing.
Gibbs Triangle,  - overpressure.
- overpressure.
The film is stretched and due to the fact that part of the liquid is gone, the surfactants rush into the free space. Gibbs triangle.
Effect of adsorption strength of bodies.
There is always an adsorption layer on the film surface, for which , then
Langmuir equation:


 into two-dimensional pressure
into two-dimensional pressure
 - analogue of the Shishkovsky equation
- analogue of the Shishkovsky equation
electrokinetic phenomena. Double electric layer (DES).
Helemholtz model. Gouy-Chapman theory.
1808 Flight

U – shaped tube, immersed in it 2 electrodes. The law of communicating vessels is violated and there is a change in the liquid level in the tube - electrokinetic phenomena.
Kinetic phenomena:
electrophoresis
Electroosmosis
Flow (flow) potential
Sedimentation potential
1 and 2 arise when a potential difference is applied, 3 and 4 the punching and sedimentation of colloidal particles cause the appearance of a potential difference.
Electroosmosis is the movement of a dispersion medium relative to a stationary dispersed phase under the action of an electric current.
electrophoresis is the movement of particles of the dispersed phase relative to a stationary dispersion medium under the action of an electric current.
P  The reason for the occurrence of electrokinetic phenomena is the spatial separation of charges and the appearance of a double electric layer.
The reason for the occurrence of electrokinetic phenomena is the spatial separation of charges and the appearance of a double electric layer.
The electric double layer is a flat capacitor, one plate is formed by potential-determining ions, the other by counterinoes. The ions are also contaminated as potential-determining co-ions are pushed into the bulk of the solution. Distance between plates  . The potential falls linearly, the potential difference
. The potential falls linearly, the potential difference  .
.
An external potential difference causes the appearance of a shear modulus  is a pair of forces per unit area acting along the surface of a solid body.
is a pair of forces per unit area acting along the surface of a solid body.

At equilibrium, the shear modulus is equal to the viscous friction modulus (  ).
).

In our conditions  ,
,

 - Helemholtz-Smalukovsky equation
- Helemholtz-Smalukovsky equation
 - linear speed displacement i phases.
- linear speed displacement i phases.
E is the electric field strength.
 - potential difference between the plates
- potential difference between the plates
 - electrophoretic mobility [m 2 /(V * s)].
- electrophoretic mobility [m 2 /(V * s)].
The Helemholtz model does not take into account the thermal motion of molecules. In reality, the distribution of ions in the double layer is more complex.
Gouy and Chapman identified the following causes of DES:
The transition of an ion from one phase to another when equilibrium is established.
Ionization of solid phase matter.
Completion of the surface by ions present in the dispersion medium.
Polarization from an external current source.
The electrical double layer has a blurred or diffuse structure. Ions tend to be evenly distributed throughout the diffuse layer.
The diffuse layer consists of counterinions, the length of the layer is determined by their kinetic energy. At a temperature tending to absolute zero, counterinoes are as close as possible to potential-determining ions.
This theory is based on two equations:
Boltzmann equation

 - work against the forces of electrostatic interaction.
- work against the forces of electrostatic interaction.
 is the bulk charge density.
is the bulk charge density.

Poisson equation 
Since the DEL thickness is much smaller than the particle size, and for a flat DEL, the derivative with respect to coordinates  and
and  is abolished.
is abolished. 
For e y with y<<1 функцию можно разложить в ряд Маклорена:

We restrict ourselves to two members of the series, then:


 - DEL thickness is the distance at which the DEL potential decreases in e once.
- DEL thickness is the distance at which the DEL potential decreases in e once.
The lower the temperature, the less  . At Т→0 – flat DES. The higher the concentration, the more I, the less
. At Т→0 – flat DES. The higher the concentration, the more I, the less  .
.





 “–” means that the potential decreases with distance. =>
“–” means that the potential decreases with distance. =>
=>

 ,
,
 - the potential decreases exponentially.
- the potential decreases exponentially.
Potential for surface charge density: 
Surface charge is a space charge with the opposite sign, integrated over distance.




=>


Where the potential decreases by 2.7 times - 
Double layer capacity 
The disadvantage of the theory is that the presence of the Helemholtz layer is not taken into account, i.e. does not take into account  , hence the errors in determining the main parameters. It also does not explain the effect of ions of different nature on the thickness of the electrical double layer.
, hence the errors in determining the main parameters. It also does not explain the effect of ions of different nature on the thickness of the electrical double layer.
Stern's theory. The structure of a colloidal micelle.
The electrical double layer consists of two parts: dense and diffuse. A dense layer is formed as a result of the interaction of potential-forming ions with specifically adsorbed ones. These ions, as a rule, are partially or completely dehydrated and can have either the same or the opposite charge to the potential-determining ions. It depends on the ratio of the energy of electrostatic interaction  and specific adsorption potential
and specific adsorption potential  . The ions of the dense layer are fixed. The other part of the ions is located in the diffuse layer; these ions are free and can move deep into the solution, i.e. from an area of higher concentration to an area of lower concentration. The total charge density consists of two parts.
. The ions of the dense layer are fixed. The other part of the ions is located in the diffuse layer; these ions are free and can move deep into the solution, i.e. from an area of higher concentration to an area of lower concentration. The total charge density consists of two parts.

 - Helmholtz layer charge
- Helmholtz layer charge
 -Diffuse layer charge
-Diffuse layer charge
The surface has a certain number of adsorption centers, each of which interacts with one counterion. The constant of such a quasi-chemical reaction is:

 , where
, where  - mole fraction of counterions in solution
- mole fraction of counterions in solution
Helmholtz distribution
The potential decreases linearly
Gouy Potential Distribution. There is no dense layer, the potential decreases exponentially from the value 
Stern distribution.
Initially, the potential decrease is linear, and then exponentially.
When an electric field is applied in the case of electrophoresis, it is not the particle of the solid phase that moves directly, but the particle of the solid phase with a layer of surrounding ions. DES repeats the shape of the particle of the dispersed phase. When a potential is applied, a part of the diffuse layer is torn off. The break line is called sliding boundary.
The potential arising at the slip boundary as a result of detachment of a part of the diffuse layer is called electrokinetic potential(Zeta potential  ).
).
A particle of a dispersed phase, with a layer of counterions surrounding it and a double electric layer, is called micelle.
Rules for writing colloidal micelles:


1-1 charging electrolyte
T is a particle of the dispersed phase.
AA is the boundary between the dense and diffuse parts.
BB is the slip boundary.
The slip boundary may or may not coincide with line AA.
The pH value at which the zeta potential is zero is called isoelectric point.
CaCl 2 + Na 2 SO 4 → CaSO 4 ↓ + 2NaCl
1. In excess of CaCl 2
CaCl 2 ↔ Ca 2+ + 2Cl -
(CaSO 4 m∙nCa 2+ 2( n-x)Cl - ) 2 x + x Cl - - record micelles.
CaSO 4 m - aggregate.
CaSO 4 m∙nCa 2+ is the core.
CaSO 4 m∙nCa 2+ 2( n-x)Cl - particle.
2. In excess of Na 2 SO 4
Na 2 SO 4 ↔2Na + + SO 4 2-
(CaSO 4 m∙nSO 4 2- 2(n-x)Na + ) 2x- 2xNa + - micelle
CaSO 4 m - aggregate.
CaSO 4 m∙nSO 4 2 + is the core.
CaSO 4 m∙nSO 4 2- 2(n-x)Na + - particle
Helemholtz-Smoluchowski equation

 - linear velocity of displacement of boundaries (in electroosmosis).
- linear velocity of displacement of boundaries (in electroosmosis).
 - potential difference on the capacitor plates (in electroosmosis).
- potential difference on the capacitor plates (in electroosmosis).


 - volumetric flow rate of the solution, S is the cross-sectional area of the cell.
- volumetric flow rate of the solution, S is the cross-sectional area of the cell.

E is the electric field strength.


 (for electroosmosis).
(for electroosmosis).
For the flow potential:

 - potential
- potential
 - membrane pressure
- membrane pressure
As a rule, the value of electrophoretic mobilities and electroosmotic mobilities is less than the calculated ones. This is due to:
Relaxation effect (during the movement of a particle of the dispersed phase, the symmetry of the ionic atmosphere is violated).
Electrophoretic braking (the occurrence of additional friction as a result of the movement of counterions).
Distortion of streamlines in the case of electrically conductive particles.
Relationship between surface tension and potential. Lippmann equation.
The formation of DEL occurs spontaneously due to the desire of the system to reduce its surface energy. In the context of constancy T and p the generalized equation of the first and second laws of thermodynamics looks like:
 (2)
(2)
(3), (1)=(3) =>
=>

 - 1st Lippmann equation.
- 1st Lippmann equation.
 is the surface charge density.
is the surface charge density.
 - differential capacitance.
- differential capacitance.
 - 2nd Lippmann equation.
- 2nd Lippmann equation.
FROM- capacity.
We solve the 1st Lippmann equation and the fundamental adsorption equation:
 ,
,


 , then
, then
 - Nernst equation
- Nernst equation
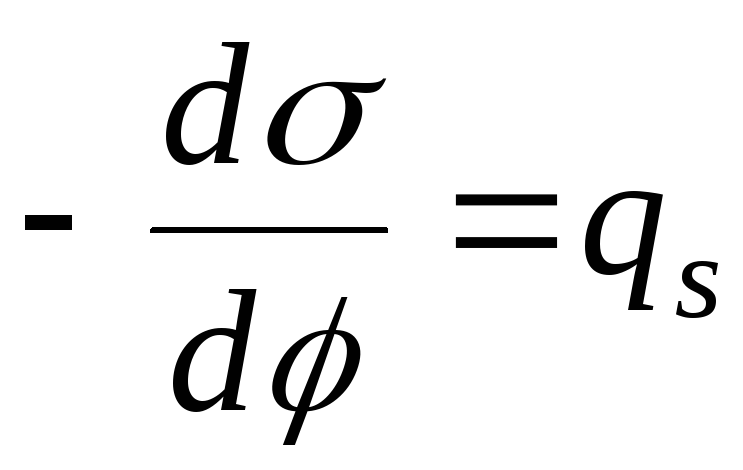 ,
,
 ,
,




 - the equation of the electrocapillary curve (ECC).
- the equation of the electrocapillary curve (ECC).
AT  :
: , but
, but 
Cationic surfactants (CSAS) reduce the cathodic branch of the ECC.
Anionic surfactants (ASS) reduce the anodic branch of the ECC.
Non-ionic surfactants (NSA) reduce the middle part of the ECC.
Stability of dispersed systems. Wedging pressure.
Dispersed systems can be divided:
Thermodynamically unstable systems can be kinetically stable due to the transition to a metastable state.
There are two types of stability:
Sedimentation stability (with respect to gravity).
Aggregative stability. (in relation to sticking)
Coagulation is the process of particles sticking together, leading to the loss of aggregative stability. Coagulation can be caused by changes in temperature, pH, stirring, ultrasound.
Distinguish coagulation:
Reversible.
Irreversible.
Coagulation proceeds with the introduction of electrolytes.
Coagulation rules:

Film- This is the part of the system located between two interfaces.
disjoining pressure occurs with a sharp decrease in the film thickness as a result of the interaction of approaching surface layers.

«-» - when the film thickness decreases, the disjoining pressure increases.
P 0 is the pressure in the bulk phase, which is a continuation of the interlayer.
P 1 is the pressure in the film.
Theory of stability. DLFO (Deryagin, Landau, Fairway, Overbeck).
According to the DLVO theory, two components are distinguished in the disjoining pressure:
electrostatic P E (positive, it is due to the forces of electrostatic repulsion). Corresponds to a decrease in the Gibbs energy with increasing film thickness.
Molecular PM (negative, due to the action of attractive forces). It is caused by the compression of the film due to chemical surface forces, the radius of action of the forces is tenths of a nm with an energy of the order of 400 kJ/mol.
Total interaction energy:

 - the system is aggregate stable
- the system is aggregate stable
 - unstable system
- unstable system
P  positive component.
positive component.
The increase is due to the increase in potential energy during compression of thin films. For thick films, the excess ion energy is compensated and is equal to the energy interaction in the bulk of the dispersion medium.
If a  (
( - film thickness,
- film thickness,  - radius of the ion) thinning of the film leads to the disappearance and reduction of molecules and ions in it with a minimum surface energy. The number of neighboring particles decreases, as a result of which the potential energy of the particles remaining in the film increases.
- radius of the ion) thinning of the film leads to the disappearance and reduction of molecules and ions in it with a minimum surface energy. The number of neighboring particles decreases, as a result of which the potential energy of the particles remaining in the film increases.


The DLVO theory considers the interaction of particles as the interaction of plates.

Particles don't interact



 - Laplace equation,
- Laplace equation,  ,
,


For weakly charged surfaces
For highly charged surfaces:

The molecular component is the interaction of two atoms:
 ~
~
Interaction of an atom with a surface:

Let's take two records:
D  To obtain the molecular component, it is necessary to sum up all the interaction energies of the atoms of the right and left plates.
To obtain the molecular component, it is necessary to sum up all the interaction energies of the atoms of the right and left plates.
where  - Hamaker's constant (takes into account the nature of interacting bodies).
- Hamaker's constant (takes into account the nature of interacting bodies).
That. the interaction energy of particles in a system can be expressed using potential curves.
I is the primary potential minimum. This is a zone of irreversible coagulation, the forces of attraction prevail.
II - zone of aggregative stability, repulsive forces prevail.
III - secondary potential minimum (or flocculation zone). Between the particles of the dispersed phase there is an electrolyte layer, and the particles can be separated and transferred to the zone of aggregative stability.

Curve 1 – the system is aggregatively stable.
Curve 2 is stable in zone I, not stable in zone II.
Curve 3 - coagulation occurred in the system.
Curve 4 - at point 4, the total energy of interaction U=0,  , this extremum point corresponds to the onset of rapid coagulation.
, this extremum point corresponds to the onset of rapid coagulation.
There are two cases:
1. Surfaces are weakly charged:
U \u003d U E + U M \u003d 0
 (1)
(1)
2)

 (2)
(2)



 - this is the thickness of the layer corresponding to the beginning of the coagulation process.
- this is the thickness of the layer corresponding to the beginning of the coagulation process.






 - for weakly charged surfaces
- for weakly charged surfaces
 then
then 
2. For highly charged surfaces:
 (1)
(1)
2)

 (2)
(2)


 (3)
(3)
 ,
,
Let's square (3)

Coagulation:
In specific adsorption, ions can be adsorbed in a superequivalent amount in such a way that the surface can change its charge. The surface is being recharged.
In the case of specific adsorption, not only ions of opposite signs, but also of one ion can be adsorbed.
If ions of the same sign as the surface are adsorbed, then in the surface layer there will be not a drop in the potential, but its growth.

Neutralization coagulation (occurs with the participation of weakly charged particles and depends not only on the charge of the coagulating electrolyte, but also on the potential at the boundary of the dense and diffuse layers).
Smoluchowski's theory of rapid coagulation.

Dependence of coagulation rate on electrolyte concentration.
I – coagulation rate is low,
II - the coagulation rate is practically proportional to the electrolyte concentration.
III - the area of rapid coagulation, the rate is practically independent of concentration.
Key points:
The initial sol is monodisperse, similar particles have a spherical shape.
All particle collisions are effective.
When two primary particles collide, a secondary particle is formed. Secondary + primary = tertiary. Primary, secondary, tertiary - multiplicity.
In terms of chemical kinetics, the coagulation process can be described by the equation:
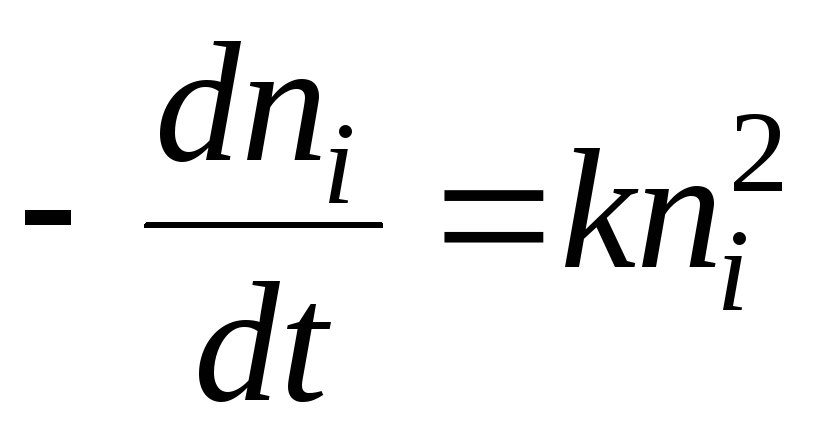
The solution will be the equation: 
 - time of half coagulation. This is the time during which the number of sol particles decreases by 2 times.
- time of half coagulation. This is the time during which the number of sol particles decreases by 2 times.

 ,
,
 ,
,
 ,
,


As the multiplicity increases, the maximum of the coagulation curves shifts towards larger values  .
.
Flaws:
Assumption of monodispersity.
The assumption about the effectiveness of all collisions.
Kuznetsova E.S. and Buryak A.K. compared the thermodynamic characteristics of the adsorption of amino acids and their associates. In this work, the influence of the structure of amino acids, their dimers and associates with eluent components on their adsorption on the surface of carbon materials was studied. A molecular-statistical calculation of the thermodynamic characteristics of adsorption (TXA) for aromatic amino acids (phenylalanine, tyrosine), heterocyclic amino acids (tryptophan) and their dimers with trifluoroacetic acid (TFA) on the surface of graphitized thermal carbon black (GTC) has been carried out. The obtained data are compared with the patterns of amino acid retention on porous graphite carbon Hypercarb under conditions of reversed-phase high-performance liquid chromatography (RP HPLC). It has been shown that TCA and amino acid retention values increase with the increase in the carbon chain of these compounds.
Shkolin A.V., and Fomkin A.A. analyzed the behavior of thermodynamic functions (differential molar isosteric heat of adsorption, entropy, enthalpy and heat capacity) of the adsorption system methane-microporous carbon adsorbent AUK depending on the parameters of adsorption equilibrium in the temperature range from 177.65 up to 393 K and pressures from 1 Pa to 6 MPa. Taking into account the influence of nonideality of the gas phase and the noninertness of the adsorbent led to the appearance of a temperature dependence of the isosteric heat of adsorption, especially in the region of high pressures of the adsorbent. For the system under study, the thermodynamic functions of the adsorption system are mainly affected by the imperfection of the gas phase. The correction for the non-inertia of the adsorbent in this range of parameters of the adsorption system is no more than 2.5%.
At the Institute of General and Inorganic Chemistry of the Academy of Sciences of the Republic of Uzbekistan Muminov S.Z. In his work, he studied changes in the surface properties and porous structure of montmorillonite upon replacement of exchangeable cations of the mineral with polyhydroxyaluminum cations. Preliminary thermal vacuum has a significant effect on the adsorption properties of polyhydroxyaluminum montmorillonite with respect to methyl alcohol. According to the series of CH3 adsorption isosteres on dehydrated sodium and modified montmorillonites, measured in a wide temperature range, the dependences of the heat of adsorption on the amount of adsorbed substance were established.
N.S. Kazbanov, A.V. Matveeva and O.K. Krasilnikova conducted a study of the adsorption of phenol from aqueous solutions with activated carbons such as FAS, PAH and carbon felt at temperatures of 293, 313 and 343K in the concentration range of 5 - 250 mmol/l. A series of samples of successively activated carbon FAS, characterized by a narrow pore size distribution, was obtained by carbonization of furfural-based polymers. PAH is a microporous polymeric activated carbon. Carbon felt is a fibrous material based on hydrated cellulose fibers. The parameters of the porous structure of the adsorbents were determined from nitrogen vapor adsorption isotherms at 77 K (ASAP-2020, Micromeritics, USA). The adsorption of solutions was studied by the ampoule method in a thermostat. The selected samples were analyzed by spectrophotometry. The analysis of the obtained isotherms of liquid-phase adsorption was carried out using the theory of volumetric filling of micropores (TOZM) according to the Dubinin-Radushkevich (DR) equation.
The effect of temperature on sorption from liquid solutions is ambiguous. On the one hand, for microporous adsorbents, the penetration of molecules into pores comparable in size to these molecules depends on the kinetic energy and, accordingly, increases with temperature. On the other hand, physical adsorption is an exothermic process and adsorption decreases with temperature. The ratio of these factors for each system determines the course of the temperature dependence of adsorption.
The uniqueness of the adsorbent - phenol system is that it has an inverse temperature dependence of adsorption isotherms, since with an increase in temperature from 293 to 313 K, the limiting value of adsorption increases, which is apparently associated with the molecular sieve effect: with an increase in temperature, phenol molecules are able to penetrate into narrower pores of carbon materials. Adsorption occurs mainly in micropores, since adsorbents have a small amount of mesopores. As the size of micropores increases, the values of limiting adsorption increase significantly, reaching 2.9 mmol/g for PAHs, 8.5 mmol/g for FAS, and 12.7 mmol/g for felt. The resulting adsorption isotherms are well described by the DD equation with the exponent equal to 2.
Interaction of polymers with liquids and gases
The processes of interaction of polymers with low molecular weight liquids play an important role in the processes of forming finished products (for example, fibers from a solution), modifying the properties (plasticization) of the material, and also in the operating conditions of these products in various liquid media. The interaction is expressed in the absorption of liquid by the polymer and is called sorption. If sorption occurs in the bulk of the polymer material, it is called absorption. If absorption occurs in the surface layers, then the process is called adsorption.
Sorption
The mechanism of adsorption is due to the presence of surface tension forces at the interfaces between media (Fig. 5.1) due to the difference in the forces of intermolecular interaction in them. This leads to the accumulation of excess energy on the surface of a substance that tends to draw in its surface molecules (molecules adsorbent) and weaker interacting molecules (molecules adsorptive) inside the volume. The amount of adsorption largely depends on the specific surface area of the adsorbent. Numerically, adsorption is expressed by the number of moles of adsorbed substance per unit mass of the adsorbent - x/m.
The study of sorption makes it possible to obtain valuable information about the structure of a polymer and the degree of packing of its molecules.
Usually, sorption processes are described using curves of the dependence of the amount of adsorbed substance on its concentration (or pressure) in the gas phase at a constant temperature (sorption isotherms, Fig. 5.2.). Here the value R/R s is the ratio of the vapor pressure of the adsorbent to the elasticity of its saturated vapor at a given temperature.

In the region of low vapor pressures, Henry's linear law is fulfilled:
where a- amount of adsorbed substance; a m- limiting adsorption proportional to the active surface of the adsorbent; p- sorbate pressure; k is the adsorption constant. On fig. 5.2 the completion of monomolecular adsorption is determined by the exit of the sorption isotherm to the shelf in the range of relative pressures 0.4 ÷ 0.5.
In the presence of polymolecular adsorption and condensation on the surface of a porous adsorbent ( R/R s > 0.6 in fig. 5.2) use the universal equation
 | (5.3) |
Thermodynamics of the adsorption process
Since, as a rule, the intermolecular interaction of adsorbent molecules is less intense than that of the adsorbent, adsorption proceeds with a decrease in the free energy of the surface (Δ F < 0) и выделением тепла (уменьшением энтальпии ΔH < 0). При равновесии процессов адсорбции и десорбции ΔF= 0. The value calculated in the course of adsorption characterizes the number and activity of groups on the surface of the adsorbent capable of reacting with the absorbent. During adsorption, the entropy of the system also decreases (Δ S < 0), поскольку молекулы абсорбтива ограничивают подвижность молекул полимера, уменьшая возможное число конформаций: ΔS = k ln ( W 2 / W 1), where is the Boltzmann constant, W 2 and W 1 - thermodynamic probability of the final and initial state of the system.
Adsorption(from Latin ad - on, at and sorbeo - I absorb), a change (usually - an increase) in the concentration of a substance near the interface ("absorption on the surface"). Cause adsorption- unsaturation of intermolecular bonds near the surface, i.e. the existence of an adsorption force field. A body that creates such a field is called an adsorbent, a substance whose molecules can be adsorbed is called an adsorbate, and an already adsorbed substance is called an adsorbate. Process reverse adsorption is called desorption.
The nature of the adsorption field is different. If adsorption is associated with van der Waals bonds, then adsorption called physical. If these are valence bonds, i.e. adsorption passes with the formation of surface chemical compounds, then adsorption called a chemical chemisorption. Important features chemisorption shows: irreversibility, high thermal effects (hundreds of kJ / mol), activated character. There are many intermediate types adsorption between physical and chemical adsorption. For example, adsorption caused by the formation of hydrogen bonds. There are also different types of physical adsorption. The occurrence of dispersion intermolecular forces of attraction is most common, due to the fact that they are approximately constant for adsorbents with a surface of any chemical nature (nonspecific adsorption). Physical adsorption can be caused by electrostatic forces (interaction between ions, dipoles or quadrupoles); wherein adsorption determined by the chemical nature of the molecules of the adsorptive (the so-called specific adsorption). The interface geometry also plays an important role. if the surface is flat, then it is adsorption open surface, in the case of a slightly or strongly curved surface - about adsorption in the pores of the adsorbent.
In theory adsorption Distinguish between statics (the adsorbent-adsorbate system is in thermodynamic equilibrium) and kinetics (there is no equilibrium).
Adsorption statics
Thermodynamics of adsorption
.Fundamentals of thermodynamics adsorption were created by J. Gibbs in the 70s. 19th century According to Gibbs, in an equilibrium two-phase system near the interface, there is some change in the local values of all extensive properties (except for the volume). However, the phases are considered homogeneous up to some geometric surface separating them. Therefore, the value of any extensive property for the system as a whole is not equal to the sum of the values of this property in homogeneous phases and . The difference is attributed to the two-dimensional surface phase associated with the separating surface. Because the surface phase has no thickness, then V0=+ and =-, where V- volume.
The presented representations allow us to reduce the fundamental thermodynamic equation to the form:
where G is the Gibbs free energy, S is the entropy, is the interfacial tension, s is the interface area, and n i- the corresponding chemical potential and the number of moles i-that component. The index indicates the value of the respective property in the surface phase. The Legendre transformation allows one to modify equation (1) for isothermal conditions:
The quantity is called the Gibbs adsorption and is denoted by the symbol G, (expressed in mol / cm 2). For a two-component system:
The position of the dividing surface can be chosen arbitrarily. In particular, the choice of this provision may satisfy the condition Г 1 =0. Such a surface is called equimolecular. For it, the designation Г 2 = Г 2 (1) is introduced. This implies the main Gibbs adsorption equation:
If the adsorbent is completely insoluble in one of the two phases, =const, and the transition from equation (2) to equation (3) does not require the condition Г 1 =0. Thus, the Gibbs adsorption is the excess of this component in a real two-phase system compared to such a system in which both phases would be strictly homogeneous up to the separating surface. In addition to the Gibbs excess quantities adsorption, plays an important role in her theory adsorption, understood as the full content of the component i in space W, which exhibits adsorption forces. Denoting the full content through a and assuming that the component i completely insoluble in one of the bulk phases, we have:
where c i-concentration i-th component in the bulk phase. For small s i:
Adsorption can occur at any interface between any two phases, in particular at the interface fluid-fluid (liquid-gas, liquid-liquid) or solid-fluid (solid-gas, solid-liquid). In fluid-fluid systems, α can be measured as a function and experimentally determined Г 2 (1) using equation (3). In the second case, to determine G 2 (1) is measured by any method n i 0 , , and concentrations of the i-th component in these volumes. From here, G is calculated i(one) . This method is called volumetric (or volumetric). With the weight (gravimetric) method, the amount is directly determined i-th component on the interface.
Adsorption isotherm
.In an equilibrium adsorption system, the parameters that determine the equilibrium are a i partial pressures R(or with i) and temperature T. They are related by the so-called thermal equation:
At adsorption individual adsorbent ( i=1) this equation takes the form:
Three special cases of the thermal equation (when T, r or a- constants) play a special role in the theory adsorption:
a=- isotherm equation adsorption,
T=- isobar equation adsorption,
R-- isostere equation adsorption.
The specific type of functions and is determined by the features of the system under consideration. If one of them, for example, is known for any value T= const, then obviously the other two also become known. In this case, it is not necessary that the analytical form of the dependencies be known. They can be given empirically as a set of values a, r and T.
In theory adsorption usually the question is about the form of the function a=(p)r, i.e. about the isotherm equation adsorption. This problem is related to thermal effects accompanying adsorption. When calculating the change in the values of the main thermodynamic functions in the case of the transition dn moles of adsorptive from the bulk phase to the surface phase in an equilibrium system at p = const, two cases are possible: in the first case, only the transformation of the adsorbate into the adsorbate is taken into account, since the adsorbent at adsorption thermodynamically unchanged and its role is to serve as a source of the adsorption field; in the second, the change in the adsorbent is also taken into account.
Since the system is in equilibrium, the chemical potentials of the adsorbate and adsorbate are the same; adsorbate entropy due to a decrease in the mobility of molecules at adsorption less than the entropy of the adsorptive. Therefore, with an inert adsorbent, the enthalpy is always negative, i.e. adsorption exothermic. Taking into account the change in the entropy of the adsorbent can change this conclusion. For example, during the sorption by polymers of substances in which the polymer swells, the entropy of the latter (due to an increase in the mobility of macromolecules) can increase so strongly that adsorption becomes endothermic. In the following, only the exothermic adsorption.
Distinguish integral, differential, isosteric and average heat adsorption. integral heat Q equal to the loss of enthalpy (at V= const - constant internal energy) when changing adsorption from a 1 before a 2(in a particular case, it can be a 1 \u003d 0): Q \u003d - (H 2 - H 1). This value is usually referred to the mass of the adsorbent and is expressed in J/kg.
Differential heat q(J / mol) is equal to the loss of enthalpy dH when it changes a on the da. It is expressed by the attitude q = - (dH/da). It's obvious that
Isosteric heat q st is taken equal to:
where is the difference between the molar volumes of the adsorbate and the adsorbate. It can be shown that  for an ideal gas adsorbent:
for an ideal gas adsorbent:
The meaning of the introduction qsi in that its determination does not require calorimetric data (such as Q and q) and it can be calculated by equation (9) from the measurement results adsorption. We also introduce the average heat Q(J/mol):
With growth a parameter Q always increasing, a q may decrease, increase or remain unchanged. With growth a with a non-uniform surface adsorption occurs in less and less active areas, which leads to a decrease q. However, in this case, the average distances between the adsorbed molecules decrease, as a result of which the forces of attraction between them increase, and q increases. The ratio between the two mentioned effects determines the course of the dependence q=f(a). At very large a repulsive forces begin to predominate in this region as well. q always decreases with growth a.
For very small surface coverages, the isotherm equation adsorption has the form of the Henry equation:
where K H is Henry's coefficient. Indeed, for very small a The adsorption layer is similar to a two-dimensional ideal gas, so its equation of state is: rt, where is the two-dimensional pressure, is the area occupied by one mole of the substance. Hence, taking into account that =-, and using equation (3), we obtain equation (12). Henry's equation requires that q was constant. For large fillings, this equation ceases to hold. Therefore, G. Freindlich (1906) proposed to describe isotherms adsorption the following empirical equation (the Freundlich equation):
where k and n- constants. This equation is often used as an interpolation formula, although for small R does not go into equation (12), and at very large R leads to an unlimited increase, which is inconsistent with experience a.
Rigorous isotherm theory adsorption was created by I. Langmuir (1914-18). The theory is based on the following. model: 1) the surface of the adsorbent is a set of energetically identical active centers on which adsorbate molecules are adsorbed (localized); 2) only one molecule is adsorbed on one center; at adsorption only one adsorption is formed. layer (monolayer); 3) adsorption on this center does not affect adsorption on other centers, i.e., interaction. adsorbed molecules can be neglected.
Langmuir model called. localized monomolecular adsorption on a uniform surface. isotherm equation adsorption corresponding to this model, maybe. obtained with the help of methods (molecular-kinetic, thermodynamic, statistical-thermodynamic). So, adsorption equilibrium can be expressed as follows. scheme:
Molecule Free. Adsorption in gas + adsorption. complex phase center (busy center)
The concentration of molecules in the gas is proportional to p, the concentration of free. centers-value ( a t - a), where and t - total number of centers, a-number of occupied centers, adsorption concentration. complexes-value adsorption Therefore, the equilibrium constant is: K p \u003d p (a t - a)/ adsorption From here we get the Langmuir equation:
where b-t. called adsorption coefficient equal to K p -1. In the region of very low pressures bp " 1 and a = (a m b)p, which corresponds to Henry's equation, in which KH= a m b. In the area of very high pressures br 1 and aa t; wherein adsorption no longer dependent on pressure. Equilibrium constant b-1 is related to the standard value of the isobaric potential of the reaction:
The Langmuir model requires that the diff. heat and entropy adsorption did not depend on the degree of surface filling.
equation (14) is a strict expression corresponding to the Langmuir model, but it is rarely justified in practice, since the model itself is idealized adsorption Doctrine of adsorption from the 20s 20th century in means. degree was built on the basis of weakening or eliminating one or another Langmuir assumption adsorption
Langmuir has already proposed a way of describing adsorption on an inhomogeneous surface (i.e., under the assumption that not all centers are the same). Combining identical centers into groups and assuming that equation (14) applies to each group, we can assume that adsorption over the entire surface is expressed by the sum of the terms of equation (14):
Assuming that the number of adsorption centers can be described by a continuous function of the distribution of the values of free. energy, Ya.B. Zel'dovich obtained from the formula (16) for the exponential function an equation of the type (13).
adsorption on inhomogeneous surfaces - a big chapter of theory adsorption Her main task-solution of the integral equation:
where f(p) - so-called. empirical isotherm adsorption, -that or another f-tion of the distribution of the number of centers on the values of free. energy,( b, p)- local isotherm adsorption, which is usually taken as the Langmuir isotherm adsorption
Many attempts have been made towards rejecting Langmuir's second assumption. adsorption On this path, the theory of polymolecular adsorption, proposed by S. Brunauer, P. Emmet and E. Teller (BET theory). The theory postulates that at a temperature below the critical temperature, each molecule adsorbed in the first layer (the heat of adsorption qi,), is the center for the molecules that form the second layer, and so on. It is assumed that the heat adsorption in all layers, except the first, is equal to the heat of condensation. This model leads to the equation:
where c = exp[(q 1 -)/RT]. equation (18) in coordinates a, p/p s corresponds to an S-curve. In coordinates p/p s ,
isotherm adsorption according to equation (18) should be linear. The slope of this straight line (usually in the range of 0.05 p/p s 0.30) and the segment cut off by it on the y-axis give the values resp. a t and With. The widespread use of the BET theory is due to the fact that its authors, in fact, considering adsorption non-localized, identify the constant a t not with the number of discrete adsorbents. centers, but with the number of adsorbate molecules in the first layer at the closest packing (at R= ps). Therefore, introducing the idea of the area occupied by one molecule in this layer, we accept:
where s- adsorbate surface area adsorption As a rule, the isotherm is measured for this adsorption nitrogen and take that for its molecule = 0.162 nm 2. A commonly performed similar calculation s according to the Langmuir model is not correct, because this method obviously only applies to non-localized adsorption
in the theory of polymolecular adsorption a great contribution was made by J. de Boer, who experimentally showed that the dependence of the average number of layers (more than the first) on all surfaces close in chemical terms. nature, from p/p s is expressed by a universal curve (the so-called t-curve). It also makes it possible to estimate the surface areas of adsorbents.
Attempts were made to take into account in the Langmuir model also the interaction. between adsorbers. molecules. So, T. Hill and J. de Boer, believing that the equation of state of adsorption. layer is a two-dimensional analogue of the van der Waals equation, we have obtained the following. isotherm equation adsorption:
where= a/a t, a and b constants of the van der Waals equation adsorption R. Fowler and E. Guggenheim, taking into account the interaction. adsorber molecules, derived the equation:
where is a constant associated with the pairwise interaction of molecules.
There is another mechanism leading to additional adsorption adsorbents below their critical. temperature on porous adsorbents at relatively high values p/p s . This is capillary condensation. If a concave adsorbate meniscus is formed in a pore, condensation begins in it at p/p s According to the Kelvin equation:
where is the surface tension of the adsorbate, V -him molar volume, r-radius of curvature of the meniscus adsorption Capillary condensation leads to a sharp rise in the isotherm adsorption In this case, the so-called is often (but not always) observed. adsorption hysteresis, i.e. adsorption mismatch. and desorbts. branches of the isotherm. As a rule, this is due to the fact that the shape of the meniscus at adsorption and desorption do not match.
Capillary condensation is used to determine the pore size of the adsorbent adsorption According to equation (22) for each value p/p s calculate the radius of curvature of the meniscus adsorption From it, given the thickness of the adsorption. layer (e.g., along the t-curve), the shape of the transition region from the layer to the meniscus and the dependence on curvature at very small r , find the linear size (effective radius r ef) of the pores filled at a given p/p s . The volume of such pores is determined by the growth adsorption at this point on the isotherm. Using the obtained data, a pore volume distribution curve along their radii is built. The method is applicable at r ef 1.5 nm. Usually the calculation is carried out by desorption. branches of the isotherm, but more strict modern. theory requires that both branches be taken into account to construct the curve.
Potential theory of adsorption and theory of volume filling of micropores. Model adsorption, fundamentally different from the Langmuir one, was proposed in 1914 by M. Polyaki. According to this model, there is potential adsorption near the surface of the adsorbent. force field decreasing with distance from the surface. As a result, the pressure of the adsorptive, which is equal to p far from the surface, increases near it and at some distance reaches the value p s at which the adsorptive condenses. The volume of the layer between the interface and the geome. place of points where p = p s , filled with liquid, which is attributed to the normal values of physical. bulk liquid properties. Reversible isothermal work e adsorption. forces, determined by the equation = RTlnp / p s, called. adsorption potential, and the whole concept is a potential theory adsorption For a given volume V adsorption layer is potential dependent on temperature (due to the independence of dispersion forces on temperature). This temperature invariance makes it possible to recalculate adsorption from one t-ry to another, although the isotherm equations adsorption it was not possible to deduce on the basis of the stated theory. The Polyani model has been widely and successfully used by many. authors, however, it contained two very vulnerable provisions: 1) the assumption that the finest adsorption. the film has normal physical values. properties of the bulk liquid (this assumption was not confirmed by experiments); 2) temperature invariance of the function =f(V), underlying the theory was approximately confirmed by experiment only for very finely porous adsorbents.
Using the potential theory, M.M. Dubinin proposed and developed the theory of volumetric filling of micro-pores (TOZM). It has been postulated that this theory only applies to microporous adsorbents. A feature of such adsorbents, in which the linear dimensions of the pores are r1 nm, is that the entire volume of their pores is "filled" with adsorbents. field. Therefore, when adsorption they are not filled in layers, but volumetrically. The value in the case under consideration is not adsorption. potential, and up to the sign of the chemical. adsorbate potential, measured from the level of chemical. potential of a normal liquid at the same temperature. The entire set of adsorbent pores is divided into three classes: micropores ( r 0.6 nm), mesopores (0.6 nm-20 nm) and macropores ( r 20 nm). adsorption in micropores occurs according to the TOZM scheme, i.e. volumetrically, in mesopores - according to the mechanism of layer-by-layer filling, completed by capillary condensation. Macropores during adsorption. equilibrium play no role.
Introducing the concept of f-tsii distribution of pore volumes on the values of chemical. adsorbate potential in them, M.M. Dubinin and L. V. Radushkevich obtained the equation for the TOZM adsorption isotherm, which is usually written in the following. form:

where n, E and a 0 -parameters ( a 0 = a at p = ps). Temperature dependence a 0:
where= -(da 0 /dT); a 0 0 = a 0 at T \u003d T 0. Options P and E practically independent of temperature. In most cases P= 2. Only for cases where the initial heats adsorption very large n > 2. To recalculate isotherms adsorption from one adsorptive to another, it is approximately assumed that E 1 /E 2 P 1 /P= and that a 01 /a 02 V 1 /V 2 , where P i- parachor, Vi- molar volume of adsorbent adsorption
Each microporous adsorbent is characterized according to the TOZM by two parameters: W- micropore volume ( W 0 = = a 0 V 0) and E 0 -characteristic. energy; W0 and E 0 refer to the standard adsorbent, usually benzene.
Using the notion that there are pores of different sizes in a real adsorbent, and introducing the distribution of values E s variance equal to F. Stekli proposed a generalization of equation (23), called the Dubinin-Stöckli equation:

where B0- constant associated with E in equation (23), and y= ![]() . Because in the adsorption the technique of naib. it was microporous adsorbents (active carbons, zeolites, finely porous xerogels) that became widespread, TOZM is used not only in physical and chemical. research, but also in engineering calculations.
. Because in the adsorption the technique of naib. it was microporous adsorbents (active carbons, zeolites, finely porous xerogels) that became widespread, TOZM is used not only in physical and chemical. research, but also in engineering calculations.
Adsorption of gas and liquid mixtures. In practice, they always deal not with an individual adsorbent, but with a mixture of gases or liquid solutions. Therefore, a generalization of the theory is required adsorption for the case of a multicomponent adsorptive adsorption In principle, one can start from any model adsorption and extend it to this case. At adsorption gas mixture, this is achieved not only by a greater complication of the equations, but also by introducing additions to them. empirical parameters associated with or with the interaction. heterogeneous molecules or, more generally, with the influence of some in-in on the coefficient. the activities of others. Only the Langmuir model allows one to obtain the isotherm equation adsorption mixtures without parameters not included in the equations for adsorption individual in-in. To do this, it suffices to take into account that during the adsorption of the kth component from a mixture i components part of the adsorption. centers can be occupied by other molecules. That's why:
When adsorption liquid solutions, regardless of their concentration, the entire surface of the adsorbent is filled with adsorption Therefore adsorption molecules of the k-th component is accompanied by the displacement of a certain number of molecules of the remaining components, i.e. adsorption is competitive.
Distinguish between molecular and ionic adsorption solutions. The first occurs when adsorption solutions of non-electrolytes, the second solution of electrolytes. Molecular adsorption, as a rule, is expressed as redundant values. Competitive nature adsorption causes the value a m.b. both positive and negative. expressing adsorption i-of that component as a f-tion of its mole fraction in solution x i-, we have that G i= O at x i= 0 and x i= 1 (a possible change in the volume of the substance in the adsorption layer is neglected). Therefore, the isotherm adsorption has one or more extremes.
isotherm equation adsorption binary solutions of non-electrolytes, reliably substantiated thermodynamically, has the form:

where the index s indicates adsorption. phase, - ( dn s 2 /dn s 1) shows how many moles of the second component are displaced by one mole of the first, the difference between the terms (standard parts) of the chemical. potential that depends only on temperature.
Main the problem of using this and a number of other isotherm equations adsorption-determination of the dependence of the coefficient. the activity of the components in the adsorption. layer from its composition adsorption The most important question in the application adsorption for separation or purification of substances - selection of a selective adsorbent in relation to this component solution adsorption
Ionic adsorption, as a rule, is not equivalent adsorption Preim are adsorbed on the surface from the electrolyte solution. cations or anions. Thanks to the electric (Coulomb) forces on the surface is formed electrical double layer.
If the adsorbent contains ions or surface func. groups capable of ionization in a given solvent, then ion exchange occurs between the adsorbent and the electrolyte solution. The adsorbent in this case is called. ion exchanger.
Adsorption kinetics
adsorption, like any real process, occurs in time. So the complete theory adsorption should contain a section on kinetics adsorption elementary act adsorption carried out almost instantly (with the exception of chemisorption). So the time dependencies adsorption are defined in the main diffusion mechanism, i.e. supply of adsorptive to the site adsorption If a adsorption on the open surface is not instantaneous, such a process occurs in the external diffusion region; while the laws of diffusion are not specific to adsorption In the case of porous adsorbents, in addition to ext. diffusion, an important role begins to play vnutr. diffusion, i.e. transfer of the adsorbent in the pores of the adsorbent in the presence of a concentration gradient in them. The mechanism of such transfer may depend on the adsorbate concentration and pore sizes.
There are molecular, Knudsen and surface (Volmer) diffusion. Molecular diffusion is carried out if the length is free. the range of molecules in the pores is less than the pore size, the Knudsen length is if this length exceeds the pore size. During surface diffusion, molecules move over the surface of the adsorbent without transition to the bulk phase. However, the values of the coefficient diffusions are not the same for different diffusion mechanisms. In many cases, it is not possible to establish experimentally exactly how diffusion occurs, and therefore the so-called. effective coefficient. diffusion, describing the process as a whole.
Main experimental material on kinetics adsorption serves the so-called. kinetic curve, i.e. f-tion \u003d a / a equal \u003d f(t) where is relative adsorption equal to the ratio of the current value of adsorption a to a equal to its value at time t. To interpret the kinetic curve in the simplest case, it is assumed that the adsorbent grain has a completely uniform porous structure in volume (this model is called quasi-homogeneous). means. improvement of the quasi-homogeneous model - the notion that each grain contains regions with larger and finer pores. Diffusion in such a grain is described by two dec. coefficients.
In the case of an open surface, taking the Langmuir model, it is easy to obtain the kinetic. the equation adsorption The rate of approach to equilibrium is the difference in speeds adsorption and desorption. Assuming, as usual in kinetics, that the rates of processes are proportional to the concentrations of reacting substances, we have:
where k ads and k dec are the rate constants respectively. adsorption and desorption. The pressure in the gas phase is assumed to be constant. When integrating this equation from t= 0 to any value t we get:
Hence, for f we have:= equal. So we finally have:
where k = k ads + k dec.
Effect of temperature on speed adsorption expressed by an equation similar to the Arrhenius equation adsorption With increasing temperature, k ads increases exponentially. Because diffusion in the pores of the adsorbent is associated with overcoming activation. barriers, the temperature dependences of k ads and k des are not the same.
Knowledge of diffusion rates is important not only for theory adsorption, but also for the calculation of prom. adsorption processes. In this case, they usually deal not with individual grains of the adsorbent, but with their layers. The kinetics of the process in the layer is expressed by very complex dependencies. At each point of the layer at a given time, the value adsorption is determined not only by the form of the isotherm equation adsorption and the laws of the kinetics of the process, but also aero- or hydrodynamic. conditions for the flow of gas or liquid around grains. The kinetics of the process in the adsorbent layer, in contrast to the kinetics in a single grain, is called. dynamics adsorption, the general scheme for solving problems of which is as follows: a system of differentials is compiled. equations in partial derivatives, taking into account the characteristics of the layer, the isotherm adsorption, diffusion characteristics (diffusion coefficient, types of mass transfer along the layer and inside the grains), aero- and hydrodynamic. flow features adsorption Initial and boundary conditions are set. The solution of this system of equations leads in principle to the values of the quantities adsorption at a given point in time at a given point in the layer. As a rule, analytical the solution can be obtained only for the simplest cases; therefore, such a problem is solved numerically with the help of a computer.
In an experimental study of the dynamics adsorption a gas or liquid stream with specified characteristics is passed through the adsorbent layer and the composition of the outgoing stream is examined as a function of time. The appearance of the absorbed substance behind the layer called. breakthrough, and the time to breakthrough - the time of the protective action. The dependence of the concentration of this component behind the layer on the time called. output curve. These curves serve as the main experimental material that makes it possible to judge the patterns of dynamics adsorption
Hardware design of adsorption processes
There are many technologies. adsorption techniques. processes. Widespread cyclic. (periodic) installations with a fixed adsorbent bed, osn. the node of which is one or several. adsorbers made in the form of hollow columns filled with granular adsorbent. A gas (or liquid) stream containing adsorbed components is passed through the adsorbent bed until breakthrough adsorption After that, the adsorbent in the adsorber is regenerated, and the gas flow is sent to another adsorber. Adsorbent regeneration includes a number of stages, of which the main one is desorption, i.e. release of previously absorbed matter from the adsorbent adsorption Desorption is carried out by heating, depressurizing the gas phase, displacement (eg, live steam), or a combination of these methods. Because times adsorption and regeneration do not coincide, select such a number of simultaneously operating and regenerated adsorbers that the whole process goes on continuously.
According to tech. and economic considerations regeneration is not brought to the end adsorption Therefore, the working capacity of the adsorbent is equal to the difference between the maximum achievable under given conditions adsorption and the amount of adsorbate remaining in the adsorbent after regeneration. As a result, the isotherms adsorption corresponding to the process in the adsorber should not be too steep.
In the described scheme, two options are possible: 1) the target product is adsorbed from the gas stream almost completely, and then it is contained in the desorbate, from where it is extracted in one way or another; 2) the target product is adsorbed worse than other components of the gas mixture, and then it is contained in the outgoing gas stream. According to the first option, for example, recuperation units at viscose factories work, capturing from exhaust gases and returning CS 2 to the cycle. The productivity of such installations reaches hundreds of thousands of m 3 of purified gas per hour; adsorbent activated carbon with not too fine micropores, i.e. coal, in which the constant E according to TOZM (see above) is 20-25 kJ / mol. This value E 0 corresponds to a not too steep isotherm, which provides good regeneration conditions. Such coals are called recovery. Desorption is carried out with live steam. To save energy, cold and hot gas flows are passed through heat exchangers.
It is very important to dry gases and liquids, such as petroleum gases before their processing or nature. gases before transportation; silica gel adsorbents or zeolites. Desorption is carried out by heating. Since the desorption of zeolite is associated with high energy costs, a combined adsorbent is used: main. mass of moisture is absorbed by easily regenerated silica gel, and deep after-drying by zeolite.
During thermal regeneration, the full cycle includes adsorption, adsorbent heating, its desorption and cooling. A large number of stages determines the low intensity and high energy intensity of the process. adsorption Therefore, the so-called. short-cycle installations, the entire cycle in which takes several. minutes. In them, gas is supplied to the adsorber under the mean. pressure, which is then released and desorption occurs. The whole process is almost isothermal (deviation from isothermality is caused only by the release of heat adsorption and absorption of heat during desorption). Cycle stages: adsorption, depressurization, desorption, pressure rise. An example is a plant with zeolite to produce oxygen-enriched air.
In installations with a moving layer of adsorbent (in the so-called hypersorbers), the latter slowly descends under the influence of gravity, is removed from the bottom. parts of the adsorber and enters the so-called. airlift, which is a vertical pipe parallel to adsorption. column. An air flow moves through this pipe from the bottom up, which raises the adsorbent grains to the top. part of the column. The processed gas flow enters the middle part of the adsorber and moves up countercurrent to the adsorbent. At the top of the column there is a continuous adsorption, at the bottom - regeneration of the adsorbent (see also adsorption cleaning).
In plants with a fluidized ("boiling") adsorbent bed, the gas flow entering the adsorber from below brings the adsorbent into suspension. This sharply increases the efficiency of mass transfer between the adsorbent and gas and reduces the duration adsorption and desorption. Such installations have high productivity. Their wide distribution is hindered by high requirements for fur. the strength of the adsorbent grains (insufficient strength causes significant losses of the adsorbent due to its abrasion and entrainment from the apparatus).
Main requirements for adsorbents: large adsorbent. capacity, i.e. they should be dispersed bodies with a large beat. surface or with a large pore volume; chem. the nature of the surface must provide effective adsorption data in-in in these conditions; chem. and thermal. durability, regenerability, availability. max. active carbons, xerogels of some oxides (silica gels, alumina gels, etc.), zeolites have become widespread; from non-porous adsorbents-tech. carbon (soot) and highly dispersed SiO 2 (aerosil, "white soot").
Fields of application of adsorption technology
On the phenomenon adsorption founded by many ways to clean the air from harmful impurities (see. gas cleaning), water (see Water treatment), as well as sugar syrups for sugar making, fruit juices and other liquids in food. prom-sti, waste lubricating oils. Removing moisture as a harmful impurity from gases and liquids using solid adsorbents is one of the important branches of adsorption. techniques (see also gas drying).
On adsorption. processes are based on the fine separation of mixtures of substances and the isolation of certain components from complex mixtures. Examples are the separation of isomers of alkanes in order to obtain normal hydrocarbons for the production of surfactants, the separation of oils in the production of motor fuels. For gas mixtures of adsorption. separation methods are used to obtain air enriched with oxygen (up to almost pure O 2); in many cases, these methods successfully compete with distillation (see. air separation).
The rapidly developing field of application of adsorbents. medical technology, where it serves to extract harmful substances from the blood (hemosorption method), etc. fiziol. liquids. The high requirements for sterility pose a very difficult task of selecting suitable adsorbents. These include specially prepared activated carbons.
Lit.: Brunauer S., Adsorption of gases and vapors, trans. from English, vol. 1, M., 1948; de Boer Ya, The dynamic nature of adsorption, trans. from English, M., 1962; Adsorption and Porosity, ed. M. M. Dubinina [et al.], M., 1976; Keliev N.V., Fundamentals of adsorption technology, 2nd ed., M., 1984; Young D.M., Crowell A.D., Physical adsorption of gases, L., 1962. M.M. Dubinin, V.V. Serpinsky.
Choose the first letter in the title of the article:
Current page: 6 (the book has a total of 19 pages) [accessible reading excerpt: 13 pages]
Font:
100% +
34. Nature of adsorption forces
The interaction between the molecules of the adsorbent with the surface of the adsorbent at the so-called. physical adsorption can be due to various reasons. Then the potential that determines the interaction of one adsorbent molecule with one atom of a nonpolar adsorptive can be expressed as follows:
θ = −Cr 6 +Br 12 ,
where r is the distance between particle centers; C is the dispersion attraction constant; B is a constant that characterizes the energy of the repulsive forces.
It is quite obvious that at relatively distant distances, attractive forces should prevail, and at close distances, repulsive forces. Also, at certain distances, these forces must be equal, which will correspond to the minimum free energy. But it is important to note that during adsorption, dispersion forces act simultaneously between each nonpolar particle.
Since the interaction energy of particles can rapidly decrease with distance, it suffices to perform summation on the nearest adsorbent atoms to determine the potential of adsorption forces. It is important that, in the case of adsorption of complex nonpolar molecules, the potential energy can be approximately calculated as the sum of all potential adsorption energies of the units of the molecule.
If the adsorbent consists of ions, then the action of already known dispersion forces can be supplemented by the action of induction forces of attraction of dipoles, which are induced in the molecules of the adsorbent by an electric field, which, in turn, is created by the ions of the adsorbent lattice.
With such an interaction, the share of inductive forces in the adsorption interaction can be proportional to the polarizability of the adsorptive molecule and the square of the field strength on this adsorbent surface.
If, on the other hand, polar adsorbent molecules are adsorbed on a polar adsorbent, then the dipoles in this case polarize the atoms of the adsorbent, i.e., as if they induce electric moments in them. Due to this influence, the inductive interaction is added to the dispersion one.
The inductive interaction itself is usually small and, depending on the dipole of the adsorptive molecule and the polarizability of the adsorbent, can reach large values. In the event that molecules are adsorbed on an adsorbent that has ions or dipoles on the surface, a so-called. interaction of ions or dipoles of the adsorbent with the electrostatic field of the adsorbent itself.
In this case, the adsorptive molecules can even orient themselves in the field of the adsorbent, and orientational Coulomb interaction occurs. It usually happens that the energies of inductive and orientational interactions are less than the energy of dispersion interactions, and therefore it is assumed that the energy of intermolecular attraction is determined by the energy of dispersion attraction.
Also, the formation of a hydrogen bond can serve as a cause of adsorption. A bond of this type can arise during adsorption on adsorbents that contain hydroxyl groups of molecules such as water, alcohols, ammonia, and amines on the surface. When a hydrogen bond is formed, the interaction energy of the adsorbent with the adsorbent can be quite large, and the heat that is released during such adsorption is much greater than the heat of adsorption of substances that are similar in shape and size of molecules, but do not form a hydrogen bond.
It is important to note that, knowing the thermodynamic description of the surface layer at the "adsorbent - adsorbent" boundary, its structure, the nature of various types of forces, the dynamics of the process, one can proceed to the study of more complex adsorption processes.
35. Adsorption as spontaneous concentration on the phase interface of substances that reduce interfacial tension
Surfactants are divided into two large groups: active and inactive substances.
Surfactants are able to accumulate in the surface layer, and in this case, positive adsorption occurs. G > 0.
Such kinds of substances must have a surface tension which, in turn, must be less than the surface tension of the solvent, otherwise the accumulation of the substance in the surface layer would be unfavorable, and must have a relatively low solubility. With sufficiently good solubility, surfactant molecules tend to leave the surface deep into the solution. Therefore, surfactants will preferentially be pushed out of the bulk of the liquid to the surface.
But with the accumulation of substances at the boundary of the solution in the molecules of these substances, which weakly interact with each other, the intermolecular interaction in the surface layer will decrease, and the surface tension will fall.
Surfactants relative to the water layer are many types of organic compounds, fatty acids with a sufficiently large hydrocarbon radical, salts of these acids (soaps), sulfonic acids and their salts, as well as various types of alcohols and amines. A characteristic feature of most molecules is their amphiphilicity: the molecule consists of two parts of a polar group and a non-polar hydrocarbon radical. Possessing a significant dipole moment and well hydrating polar group can determine the affinity of the surfactant for the aqueous environment. But the hydrocarbon radical is the cause that lowers the solubility of these compounds.
Surface inactive surfactants- these types of substances, tending to leave the surface of the liquid into its volume, as a result, the so-called. negative adsorption G < 0. Поверностно-инактивные вещества также обладают значительным поверхностным натяжением, значительно большим, чем натяжение у растворителя (иначе эти вещества способны самопроизвольно накапливаться в поверхностном слое), также обладают высокой растворимостью, что способствует их стремлению уйти с поверхности жидкости в объем. Взаимодействие между молекулами поверхностно-инактивного вещества и растворителя всегда больше, чем взаимодействие между самими молекулами растворителя, поэтому они и стремятся перейти в объем раствора. Surface-inactive substances in relation to water are many inorganic electrolytes: acids, alkalis, salts. Molecules of surface-inactive substances do not have a hydrophobic part and can decompose in water into highly hydrating ions.
Examples surface-inactive substances are also some organic compounds in which the non-polar part of the molecule is absent or very small. These substances include formic, aminoacetic acids.
In non-aqueous solvents, inorganic electrolytes are also capable of increasing the surface tension, and this depends on the solvent.
For example, when sodium iodide is introduced into methanol, the surface tension greatly increases; for ethanol, the surface tension is approximately 2 times greater. The surface activity of substances can depend not only on the nature of the substance, but also on the properties of the solvent. If any solvent has a high surface tension, then this solute may exhibit significant surface activity.
36. Adsorption theories
Let us consider the most common adsorption theories that describe individual types of adsorption on the “solid-gas” or “solid-solution” interface.
The theory of monomolecular adsorption by I. Langmuir.
1. Adsorption is localized and is caused by forces close to chemical ones.
2. Adsorption occurs only on active centers - protrusions or depressions on the surface of the adsorbent, characterized by the presence of free valences. Active centers are considered independent and identical.
3. Each active center is able to interact with only one adsorbate molecule; only one layer of adsorbed molecules can form on the surface.
4. The process of adsorption is reversible and equilibrium; the adsorbed molecule is retained by the active center for some time, after which it is desorbed; After some time, a dynamic equilibrium is established.
The maximum possible adsorption value G o is achieved under the condition that all active centers are occupied by adsorbate molecules. Monomolecular adsorption isotherm equation relating the adsorption value G with adsorbate concentration FROM, looks like:
where b- a constant for a given pair of "adsorbent - adsorbate" value (the ratio of the rate constants of desorption and adsorption), numerically equal to the concentration of the adsorbate, at which half of the active centers are occupied.

The graph of the Langmuir adsorption isotherm is shown in Figure 2. The constant b we define graphically by drawing a tangent to the adsorption isotherm at the point FROM= 0. When describing the process of adsorption of gases in the equation, the concentration can be replaced by a proportional value of the partial pressure. Theory of monomolecular adsorption I. Langmuir applicable to describe the processes of adsorption of gases and dissolved substances at low pressures (concentrations) of the adsorbate.
Polanyi's theory of polymolecular adsorption describes s-shaped adsorption isotherms, the shape of which indicates the possible interaction of adsorbed molecules with the adsorbate.
1. Adsorption is caused by physical forces.
2. The surface of the adsorbent is homogeneous, there are no active centers; adsorption forces form a continuous force field near the surface of the adsorbent.
3. Adsorption forces act at a distance greater than the size of the adsorbate molecule, i.e., there is a certain adsorption volume near the surface of the adsorbent, which is filled with adsorbate molecules during adsorption.
4. The attraction of an adsorbate molecule by the adsorbent surface does not depend on the presence of other molecules in the adsorption volume, as a result of which polymolecular adsorption is possible.
5. Adsorption forces do not depend on temperature, and, therefore, with a change in temperature, the adsorption volume does not change.
Freundlich equation. The surface of the adsorbent is inhomogeneous, interaction occurs between adsorbed particles, active centers are not completely independent of each other. G. Freindlich suggested that the number of moles of adsorbed gas or solute per unit mass of the adsorbent (the so-called specific adsorption X/m), should be proportional to the equilibrium pressure (for gas) or the equilibrium concentration (for substances adsorbed from solution) of the adsorbent raised to a certain power, which is always less than unity:
x / m = aP n x / m = aC n.
exponents n and proportionality factor a determined experimentally.
37. Thermodynamics of the adsorption process. Gibbs adsorption equation
To study the phenomenon of adsorption at the "solution - gas" boundary, it is necessary to establish a relationship between the excess of the adsorbed substance in the layer on the surface ( G), surfactant concentration in solution ( With) and surface tension ( σ ) at the “solution-gas” phase boundary. It is more expedient to consider the phenomena from the thermodynamic standpoint and relate the adsorption of a solute to a change in the free energy of the surface or its surface tension. This connection was made W. Gibbs in 1876, which was named "Gibbs adsorption equation":
G = – With / RT x dσ/dc.
You can still imagine Gibbs Equation, based on thermodynamics, using isobaric-isothermal potential G, chemical potentials μ 1 and μ 2 , and also using n 1 and n 2 the number of moles of the components. Having analyzed it taking into account the entropy S, volume V and pressure P, we can write the following equation:
dG=– SDT+VdP+σds+ μ 1 d n 1 + μ 2 days 2 .
We equate it to zero, and taking into account constant temperature and pressure, it simplifies into an equation of the form:
sd σ + n 1 d μ 1 + n2d μ 1 = 0.
Taking into account the fact that for dilute solutions the chemical potential of the second component is expressed as follows:
μ 2 = μ 2 0 +RT ln c,
and given that the temperature is constant
dμ 2 =rtdnc,
substituting this equation into
![]()
we obtain the desired Gibbs adsorption equation. Based on the equation, it can be seen that if the surface tension σ increases with concentration With, then the concentration of the dissolved substance on the surface layer is less than in the volume of the solution (the so-called negative adsorption), and if the surface tension σ decreases with increasing concentration With, then the concentration in the layer is greater than in the volume (positive adsorption), and, finally, if σ does not depend on With, then the concentration of the substance in the layer on the surface and in the volume is the same. The Gibbs equation was derived using thermodynamics. It is difficult to verify this equation in practice, which is due to the complexity of determining the concentration of a dissolved substance in a layered surface. Experienced B. McBen found that a very thin layer of liquid was cut off from the surface of the solution using the device. Further determination of all parameters of the Gibbs equation showed that the experimentally found values of adsorption coincided with the values calculated using the Gibbs equation within the experimental error. Due to the homogeneity and smoothness of the surface of any liquid, when studying adsorption on its surface, the usual ideas about active centers are completely inapplicable. At the critical temperature, the difference between the adjacent phases disappears, and the surface tension, as a rule, becomes equal to zero. The adsorption of gases and vapors has such a large practical application that in the literature, especially in the technical one, you can find this concept, which is used only in relation to processes on the surface of solids.
This concept, as well as the most general patterns of adsorption, as the considered Gibbs equation, is applicable to all phase boundaries. Using the Gibbs equation and all the provisions arising from it, having determined the value of Г, it is possible to construct an adsorption isotherm.
38. Peculiarities of adsorption on microporous materials. Polan's potential theory. Adsorption potential
Glade's theory considers non-localized physical adsorption, which is directly due to van der Waals forces between the adsorbent and adsorbate (this can be considered the first position). The second position of this theory is the concept of the force (or potential) field of the adsorbent, which extends over a considerable distance from the surface; the adsorption layer that appears in this field is polymolecular. If we consider the adsorption of gases, then the density of this layer decreases along a certain normal from the surface. If we consider vapor adsorption, then a liquid layer of a certain thickness is formed on the surface. The field in Polanyi's theory is considered as a series of equipotential surfaces, each surface corresponds to a certain value of the potential ε , and each subsequent surface will be smaller than the previous one. Each such surface in space cuts out layers of a certain volume, designated as v i. The task of Polanyi's theory is to find the transition from the usual coordinates of the isotherm ( x, p) to field parameters ε i and v i, with further establishment of the connection between these main parameters. The first part of the problem, which Polanyi laid down, is rather complicated, and in many cases cannot have definite solutions, but for the case of vapor adsorption, this part of the problem is solved in the first approximation very simply. For a liquid adsorption layer, the filled part of the volume will be equal to:
v i \u003d x (M / d),
where d is the density of the substance in the liquid state.
In his theory, M. Polyany introduces another provision about the absence of the so-called. field screening in the process of adsorption, the value ε in this theory of space is a constant value (something like a gravitational potential) regardless of whether there are certain adsorbate molecules between a given point and a solid surface, or whether all space is free. Polyani introduces the concept adsorption potential ε , which is the isothermal work of compressing the vapor when it is transferred from the equilibrium pressure R in the bulk phase far from the surface to the region of the surface layer with saturated vapor pressure p 0 then the expression for determining the potential will look like:
ε = RT ln R 0 / R.
With the help of such an equation, one can go from the coordinates x, p to the coordinates ε and v and get a curve, which is called "characteristic". Polanyi discovered in his experiments that such curves, constructed from the experimental data of the obtained isotherms, have the following property: they are invariant with respect to T, or, in other words, all curves of this type can lie on one curve ε −ε .
M. Polyany accepted this position as a postulate, i.e.:
This property of Polyani is of great practical importance; it can construct a family of isotherms from one experimental adsorption isotherm.
Polanyi's theory does not give an analytic expression for the isotherm or the function of potential versus volume, but allows one to calculate the coordinate for any given temperature if at least one isotherm is known. This result is very important for technological calculations, because for similar gases on the same adsorbent, the adsorption curves can turn out to be close to each other and can in many cases be superimposed.
39. Characteristic curve of adsorption. Temperature invariance and affinity of characteristic curves
The force field that appears at the surface of the adsorbent can be similar in many respects to the gravitational field. In the adsorption field, potential surfaces can be represented, i.e., surfaces for which the same adsorption potential is characteristic. Under the concept of adsorption potential θ should be understood as nothing more than the work done against the forces of adsorption when moving 1 mole of the adsorbate from a certain point in the field to a certain gas phase. The maximum adsorption potential will exist at the “adsorbent – adsorption volume” boundary. But at the boundary "volume - gas phase" (this is where the action of adsorption forces ends), the adsorption potential must be equal to zero. The change in the adsorption potential with a change in the adsorption volume can be represented in the form of curves. This was first done by M. Polyani. Such types of curves do not depend on temperature and can be characteristic of each particular adsorbent; such types of curves are usually called characteristic curves of adsorption. The theory of polymolecular adsorption assumes that the equation of state of the gas applies to the amount of adsorption. Consequently, the isotherms that characterize the dependence of the density of the adsorbate on the volume for different temperatures resemble the isotherms of the dependence of the pressure on the volume. At low temperatures, adsorption forces on the surface can cause the vapor to condense into a liquid of a certain density. At temperatures lower than the critical one, during condensation, the entire adsorption volume will be filled with liquid. In this case, the adsorption curve will run almost parallel to the abscissa axis, which is related to the low compressibility of the liquid. Then the adsorption curve at the “volume – gas phase” boundary drops sharply down, and, accordingly, the density of the adsorbate reaches the value of a certain density of the gas phase. At temperatures higher than critical, the adsorbent may behave like an ideal gas, and the graph will be expressed as an isotherm of the dependence for an ideal gas, provided that pV = RT. Under such conditions, the adsorbed gas will have a maximum density at the very surface of the adsorbent and have a minimum density in the immediate vicinity of the gas phase. Moreover, in this case it is important to note that the density of the adsorbate in the adsorption layer nowhere reaches the density of the liquid itself. And if the temperature is very close to critical, the dependence of density on volume will be expressed by a curve close in appearance to the isotherm, which is described van der Waals equation. In this scenario, part of the adsorbed substance will be in the adsorbed volume in a liquid state, and part of the adsorbed substance will be in a gaseous state. Then the curve will decrease most sharply in the section that corresponds to the transition from liquid to gas. If a characteristic curve is constructed from the experimental adsorption isotherm of one of the adsorptives, and knowing the corresponding affinity coefficients for some other adsorptive, one can find the adsorption isotherm and construct it for another adsorptive. The potential theory of adsorption makes it possible to calculate different adsorption isotherms of different vapors on the same adsorbent, moreover, using the characteristic curve obtained from the adsorption isotherm of one vapor, since the ratio of the adsorption potential does not depend on the adsorption volumes.
affinity(from Latin affinis - “related”) - affinity chromatography. The method of purification and separation of proteins is based on their selective interaction with a ligand covalently bound to an inert carrier (affinity chromatography). Measuring the affinity of a toxicant for a receptor, in fact, is an experimental study of the relationship between the amount of a substance added to the incubation medium and the amount of the toxicant-receptor complex formed as a result of the interaction.
